Mate Pair Sequencing
Introduction to Mate Pair Sequencing
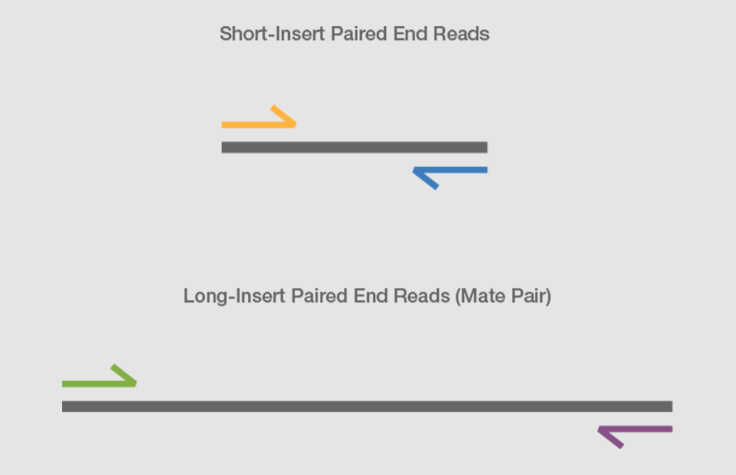
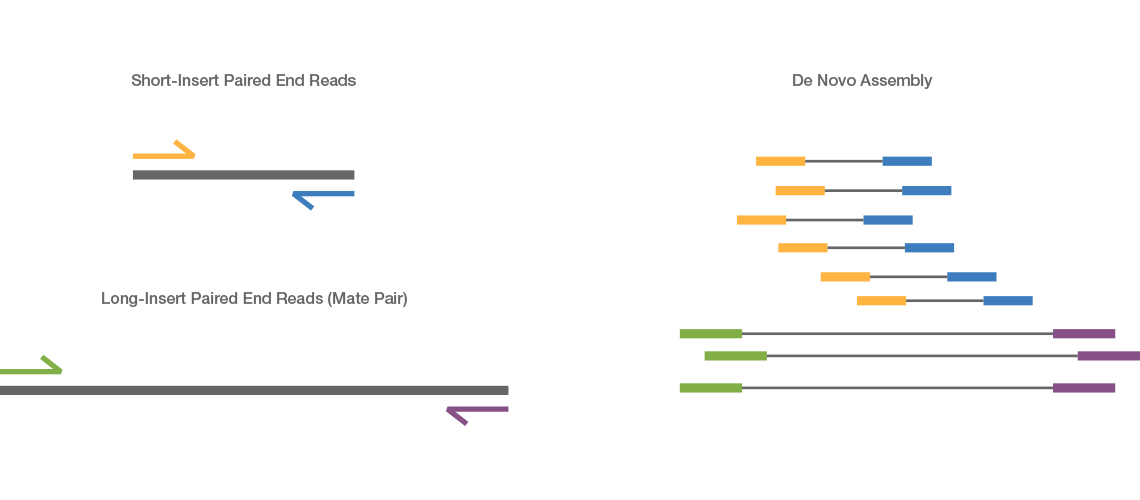
Mate pair sequencing involves generating long-insert paired-end DNA libraries useful for a number of sequencing applications, including:
- De novo sequencing
- Genome finishing
- Structural variant detection
- Identification of complex genomic rearrangements
Combining data generated from mate pair library sequencing with that from short-insert paired-end reads provides a powerful combination of read lengths for maximal sequencing coverage across the genome.
Mate Pair Library Preparation Process
Following DNA fragmentation, the DNA fragments are end-repaired with labeled dNTPs. The DNA fragments are circularized, and non-circularized DNA is removed by digestion. Circular DNA is fragmented, and the labeled fragments (corresponding to the ends of the original DNA ligated together) are affinity-purified. Purified fragments are end-repaired and ligated to Illumina paired-end sequencing adapters.
Additional sequences complementary to the flow cell oligonucleotides are added to the adapter sequence with tailed PCR primers. The final prepared libraries consist of short fragments made up of two DNA segments that were originally separated by several kilobases. These libraries are ready for paired-end cluster generation, followed by sequencing utilizing an Illumina next-generation sequencing (NGS) system.
Explore the NovaSeq 6000 System
Scalable throughput and flexibility for virtually any genome, sequencing method, and scale of project.
Learn More
Highlights of Mate Pair Sequencing
High Genomic Diversity
Efficient protocol enables the highest genomic diversity of any next-generation platform.
User-Friendly Workflow
Simple workflow with limited hands-on time and multiple stopping points.
Low DNA Input Requirements
Requires as little as 1 μg of starting material.
Mate Pair Sequencing for Kiwi Genome Assembly
See how researchers used mate pair sequencing for de novo assembly of the kiwi bird genome.
Interested in receiving newsletters, case studies, and information from Illumina based on your area of interest? Sign up now.
The Nextera Mate Pair Library Kit has been discontinued. There will not be a replacement kit.