Abstract
- Constellation technology utilizes a highly simplified NGS workflow that enables on-flow-cell library prep that completely eliminates standard library prep prior to sequencing
- Standard cluster generation and SBS sequencing is combined with cluster proximity information in DRAGEN algorithms to unlock long-distance information
- Early testing demonstrates enhanced mapping of challenging genomic regions, ultra-long phasing, and improved detection of large structural rearrangements
- The first commercialized product, expected to launch in 2026, will enable cost-effective, comprehensive, human WGS accessible with existing NovaSeq X systems
Introduction
Next-generation sequencing methods, primarily sequencing-by-synthesis (SBS), have advanced significantly over the past 25 years, making it a widely used technique in genomic applications. While Illumina short-read whole-genome sequencing (srWGS) achieves highly accurate coverage over most of the human genome,1 specific regions of the genome have remained challenging to resolve. Additionally, some classes of genomic variation, including large structural rearrangements, are difficult to detect using standard short reads. Longer read lengths have a demonstrated ability to address some of these challenges but are comparatively costly and challenging to scale, with high input amounts and stringent input requirements.
Here, we introduce constellation mapped read technology, an innovative approach that leverages on-flow-cell library prep and informatics that utilize proximity information from clusters in neighboring nanowells to generate long-range genomic insights using standard SBS sequencing. Constellation technology maintains the accuracy, depth of coverage, and scalability of standard SBS while adding the phasing, enhanced mappability, and improved structural variant detection often associated with long-read methods. This novel approach provides a powerful, accessible solution for comprehensive whole-genome analysis.
Consult the end of this post for a glossary of terms.
A novel technology
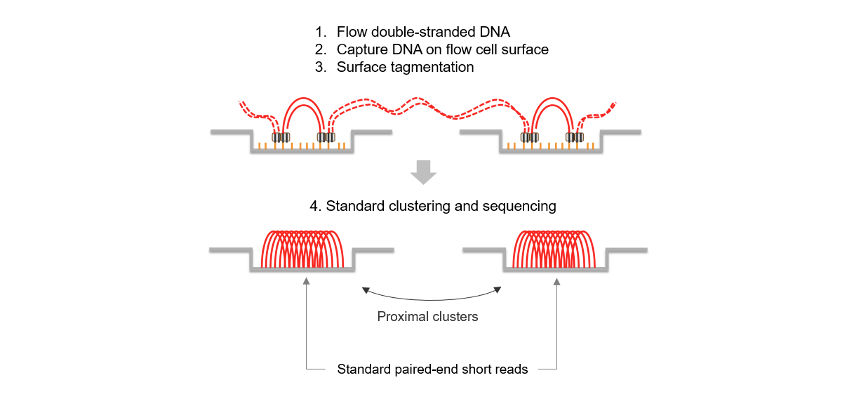
Constellation technology is unlike other methods that generate long-distance information from standard short-read sequencing (Hi-C or linked-reads, for example). Constellation tech eliminates manual library prep by applying extracted DNA directly to the flow cell surface, where surface-bound transposomes perform tagmentation in situ. This on-flow-cell library prep ensures that adjacent regions in a sample’s genome remain physically proximal on the flow cell. Instead of relying on complex tagging, molecular barcodes, or long contiguous reads, constellation tech leverages the spatial proximity of neighboring clusters to unlock long-range genomic information from a sample’s genome using standard SBS sequencing, resulting in:
- Improved mapping and variant calling in difficult-to-map regions
- Ultra-long phasing, up to several megabases
- Improved calling of large (> 50 bp) structural rearrangements
How it works
Highly simplified, on-flow-cell workflow
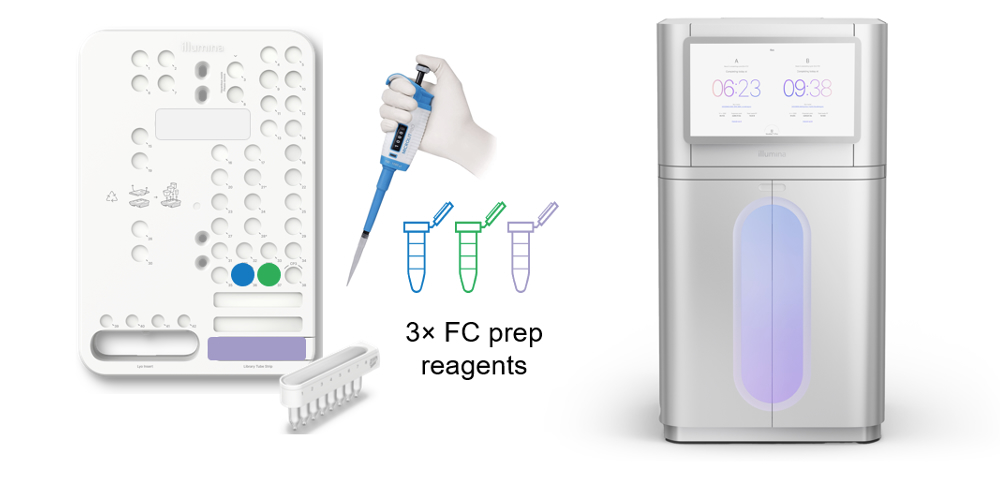
The constellation workflow begins with a novel, on-flow-cell library prep that uses the low DNA inputs characteristic of transposome-based library prep workflows and the high sequencing quality of the NovaSeq™ X series. The experimental workflow requires no modifications to the sequencing instrument—only a custom sequencing recipe, making it accessible to a large existing install base.
The constellation experimental workflow:
- Add extracted DNA template to library strip tube
- Add specialized reagents to the custom primer wells of the sequencing cartridge
- Load consumables and initiate run
Constellation read mapping and proximity analysis
The benefits of constellation technology go far beyond workflow simplification—using proximity information, reads from neighboring clusters are reconstructed into an interspersed version of the original DNA template molecule. This is demonstrated in Figure 3, where each node represents a read pair deriving from a flow cell cluster, and the lines between them indicate predicted connections between them based on a combination of flow cell and genomic proximity. Figure 3 further makes this interspersed representation explicit, denoting the genomic distance between reads coming from the same original template molecule, with template lengths beyond 300 kb. Connections are derived from a proximity model that provides a Phred-scaled quality score describing the probability that two reads have landed with a certain flow cell displacement and within a given genomic distance by chance. The higher the score, the more likely it is that two reads were derived from the same template molecule. This property is unique to constellation technology and is not observed in any other NGS assay. Reads derived from the same template molecule also share the same haplotype. This combination of Phred-scaled proximal quality and general proximity properties is used to assign reads to challenging-to-map regions, extract phase information, and call variants using DRAGEN secondary analysis.
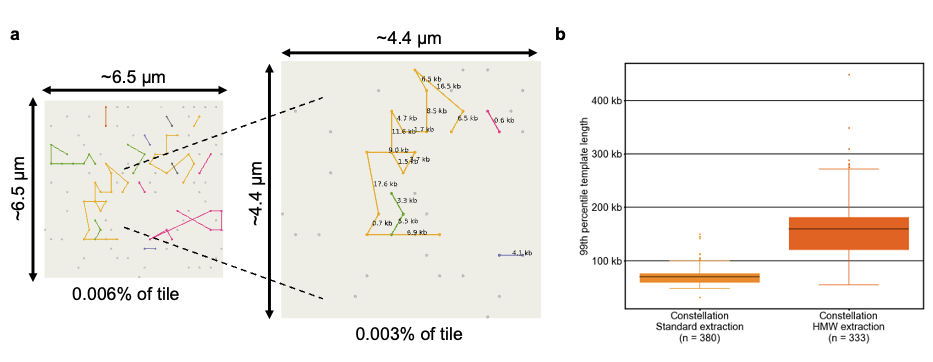
Figure 3a: Clusters generated from the flow cell are organized into distinct templates, visually represented in different colors. Within each template, the clusters are sequentially arranged based on their genomic coordinates. The connections between paired reads are annotated with their corresponding genomic distances.
Figure3b: The box plot shows the range of 99th percentile template sizes from standard and HMW extractions
The constellation advantage
Improved performance in difficult-to-map regions
In limited regions of the genome, uniquely mapping standard short reads is challenging due to high homology or other repetitive context, which makes it difficult to distinguish among multiple candidate mapping positions. Constellation read mapping uses proximity information from neighboring clusters that do uniquely map to assign reads to the correct genomic location.
The application of proximity information results in more confident mapping and comprehensive coverage of the genome, including difficult-to-map, medically relevant genes such as STRC and PMS2 (Figure 5 and Figure 6)
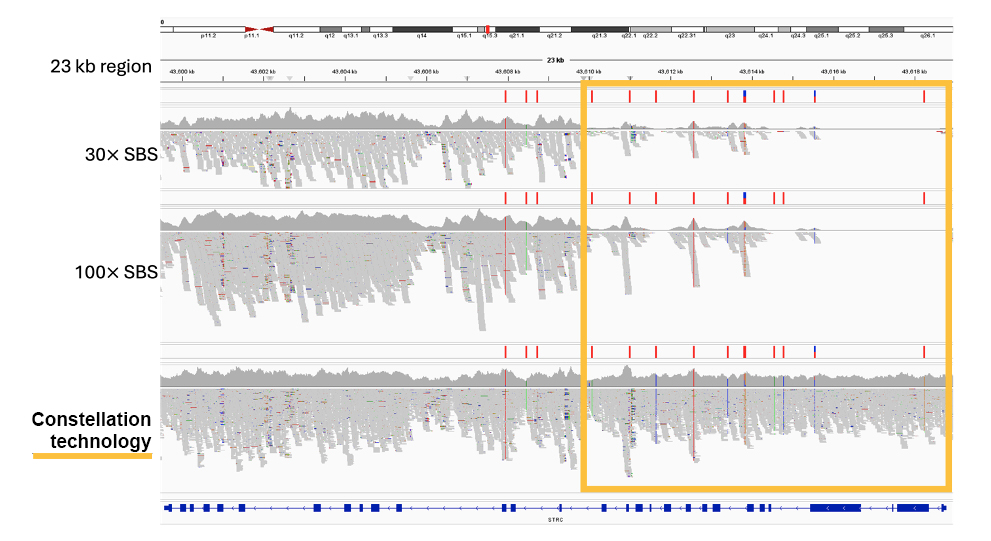
Figure 5: Recovery of coverage in the STRC gene with constellation technology. STRC has a pseudogene, STRCP1, with > 99% sequence identity, making mapping challenging with standard whole-genome library prep. Some mutations in STRC are associated with pediatric nonsyndromic hearing loss.
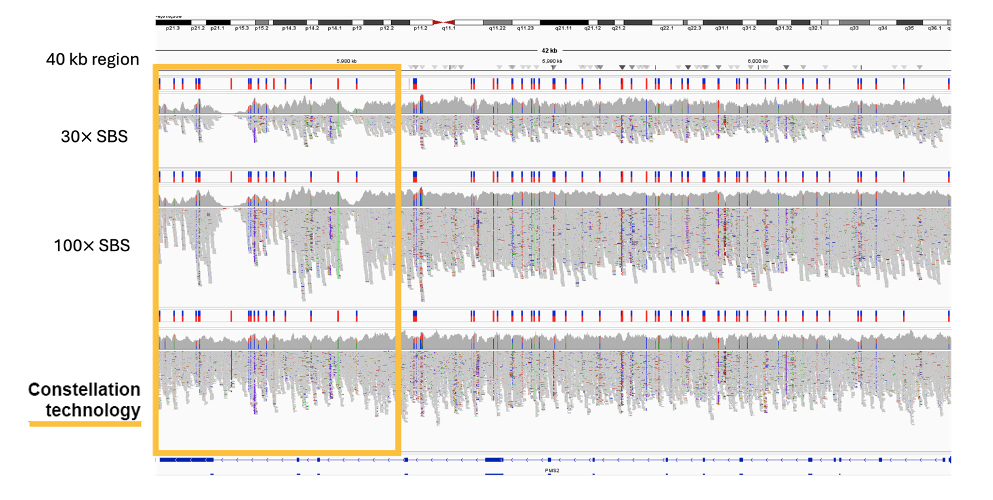
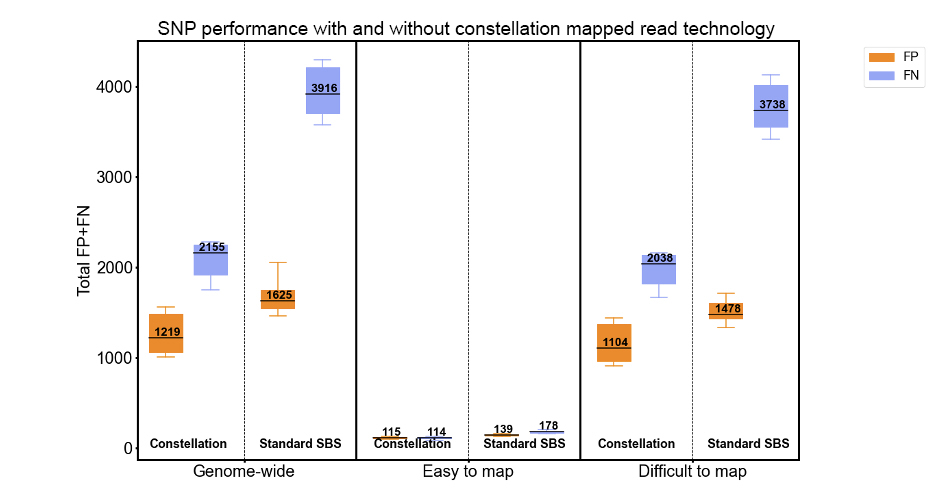
Figure 7: SNP variant calling performance assessed against NIST Genome in a Bottle v4.2.1 truth set using rtgeval for 11 HG002 runs of constellation read mapping. Difficult-to-map and easy-to-map stratifications are defined by Genome in a Bottle’s v3.3 genome stratifications as GRCh38_alldifficultregions.bed.gz and GRCh38_notinalldifficultregions.bed.gz, respectively.
Ultra-long phasing
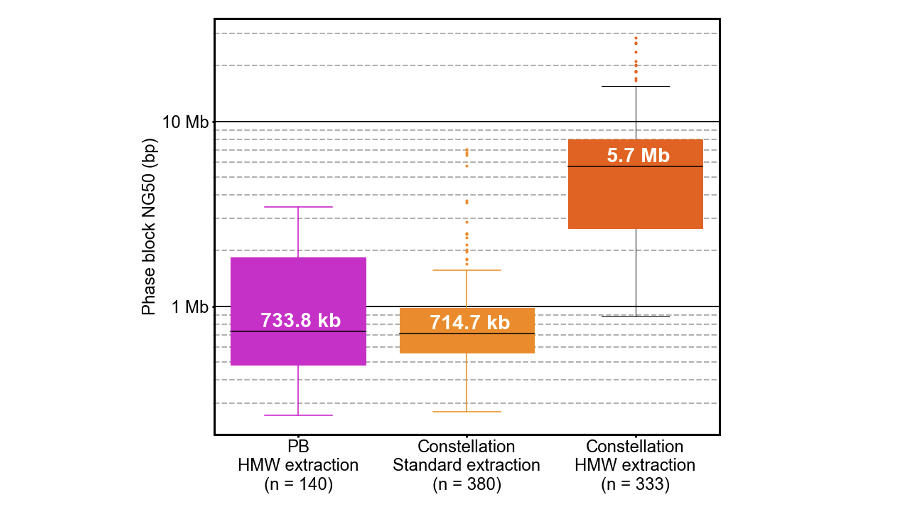
Phased sequencing enables greater insight by defining haplotypes and enabling identification of compound heterozygotes. Phasing with constellation technology is especially powerful since its capabilities are defined solely by the native DNA template length captured on the flow cell, not read length, and currently extends from hundreds of kilobases up to several megabases. High molecular weight (HMW) extraction methods that preserve larger templates are shown to contribute to larger phase blocks.
Constellation phase block NG50s are ~715 kb with standard DNA extractions and ~5.7 Mb with HMW DNA extractions (Figure 8). Initial testing demonstrates that constellation tech fully phases a median of ~85% of all genes with standard DNA extraction and ~95% of genes with HMW DNA extraction. Additionally, constellation tech phases ~98% of all heterozygous SNVs in both standard and HMW DNA extractions.
Improved structural variant calling
Constellation technology has the added benefit of improved structural variant (> 50 bp) calling. Using DRAGEN v4.3 secondary analysis, constellation tech shows a dramatic improvement in SV recall, from 51.5% with standard SBS to 87.8% (Figure 9).
With further development of constellation tech and tailored variant calling methods, we anticipate further improvements in read mapping and both small and large variant calling.
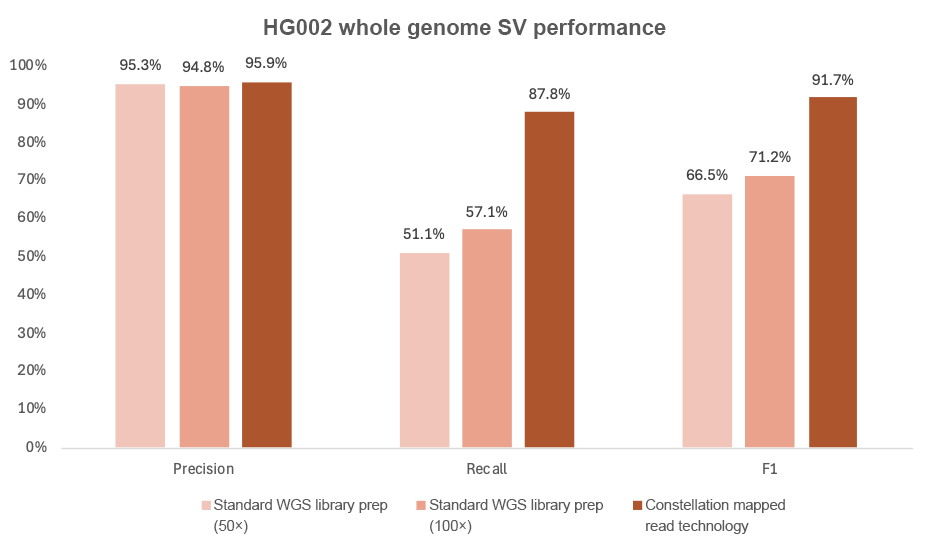
Figure 9: Constellation technology SV performance as compared to DRAGEN v4.3 at 50× and 100×. The analysis uses the Genome in a Bottle T2T-Q100 HG002 SV v1.1 truth set with the SV confident BED file. Benchmarking was performed in accordance with Genome in a Bottle guidance for structural variant benchmarking, using “bench” and “refine” commands from Truvari v4.2.2.
Novel visualization of genomic structure
Constellation mapped read technology’s ability to resolve large structural rearrangements includes novel capabilities that extend beyond traditional variant calling performance benchmarks. By extracting information about reads from proximal clusters between any pair of regions of the genome, high-resolution visual representations of genome structure maps—termed “colocation plots”—become possible.
These maps are generated by dividing the genome into bins and determining the number of reads in neighboring clusters for each possible pair of genomic bins. A high number of reads from neighboring clusters in a pair of bins occurs almost exclusively when those bins are in close genomic proximity. In a region with no structural variants, genomic bins that are nearby in a given reference genome are also nearby in a sample, and so appear as a diagonal line in a colocation plot. When a structural variant is present within a region, genomic bins that are nearby in the reference genome are no longer nearby in the sample, and so exhibit a variety of off-diagonal signals.
Figure 10 provides examples of these maps for a region overlapping the F8 gene on chromosome X, with Figure 10a showing a sample with no SV present and Figure 10b showing a sample with an intron 22 inversion.
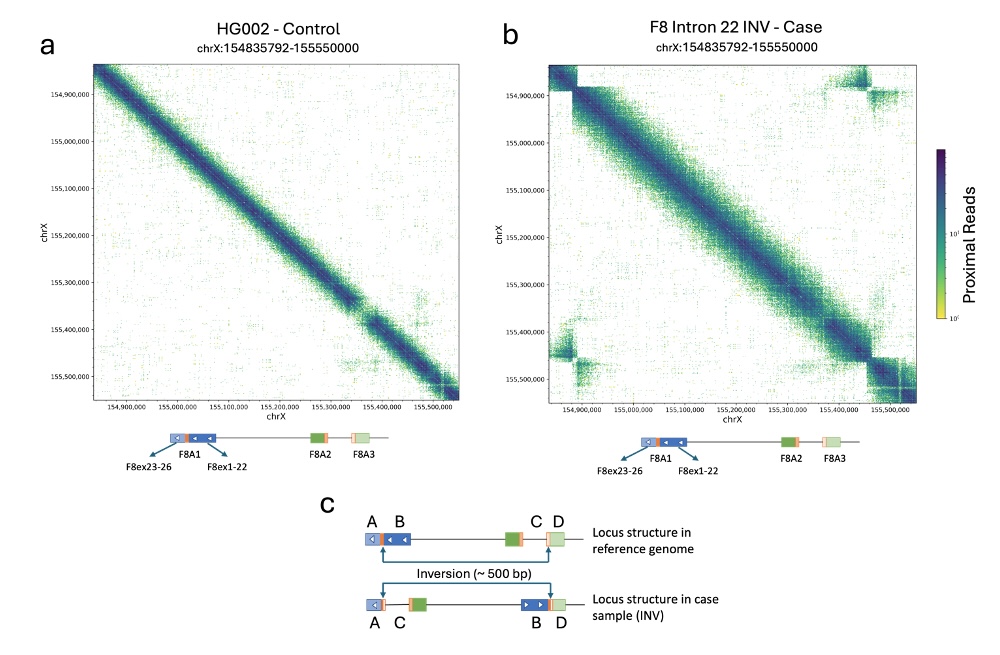
Figure 10a: Colocation plots for the F8 gene locus on chromosome X for the HG002 cell line
Figure 10b: Colocation plot for the F8 gene locus on chromosome X for a DNA sample with intron 22 inversion. Accumulation of off-diagonal signal between F8 exons 23–26 with the region upstream of F8A3 as well as between F8 exons 1–22 with the region downstream of F8A3 indicate an inversion with boundaries in intron 22 and F8A3.
Figure 10c: Diagram representing the structure of the F8 gene locus in the reference genome as well as in the sample with the pathogenic inversion. When inversion is present, novel signal is seen between regions A (F8 exons 23–26) and C (upstream of F8A3) as well as between regions B (F8 exons 1–22) and D (downstream of F8A3).
A lack of signal in the diagonal can also be observed in the sample carrying the inversion, indicating that the regions on either side of the inversion boundary are distal from each other in the case sample.
The inversion event displayed in Figure 10b has one boundary in a segmental duplication within F8 intron 22, and the other boundary in a corresponding segmental duplication in F8A3 (~500 kb away). The segmental duplications are ~10 kb in length, are in reverse orientation, and have > 99.7% sequence similarity. These characteristics make the inversion undetectable with standard short-read sequencing.
Colocation plots enable both the detection and visualization of complex balanced and unbalanced structural rearrangements, even when the event boundaries occur in difficult-to-map regions of the genome.
Conclusion and next steps
This is just the beginning
Constellation mapped read technology is a powerful new foundational technology with broad capabilities—here we demonstrate some of its benefits for human genome sequencing, however multiple future applications are under evaluation. The first commercially available product based on constellation technology is slated for the first half of 2026 and will leverage existing NovaSeq X Systems to create an accessible, cost-effective solution for comprehensive human WGS.
Follow this link to view the ASHG 2024 presentation from Illumina Chief Technology Officer Steve Barnard and Broad Clinical Labs Chief Scientific Officer Niall Lennon demonstrating early results.
Sign up below to stay updated on the future developments of constellation mapped read technology and future products.
To find out more
Reference
- Behera S, Catreux S, Rossi M, et al. Comprehensive genome analysis and variant detection at scale using DRAGEN. Nat Biotechnol. Published online October 25, 2024:1-15. doi:10.1038/s41587-024-02382-1
Glossary
| Term | Definition |
|---|---|
Template molecule |
A large contiguous double-stranded DNA molecule that has been extracted from a sample |
Standard WGS |
Whole-genome sequencing performed with manual library preparation and with standard SBS sequencing |
Tagmentation |
The process of cutting a fragment of DNA and adding an adapter sequence (tagging) using a transposome |
Transposomes |
DNA transposase complexes that exist as a dimer |
Cluster |
An amplified spot of DNA on a flow cell, that will be sequenced |
Proximal clusters |
Clusters that are physically near each other on the flow cell. |
Phase block NG50 |
The length of the phase block once 50% of the target region (genome or other) has been phased. Note that a technology that is unable to phase 50% of a given target region will have an NG50 of zero bp. |
Percent of genes fully phased |
The percentage of genic regions from a given source (for example, NCBI RefSeq, ENCODE, MANE) that are completely contained within a single phasing block. |
Percent heterozygous variants phased |
The percentage of phased heterozygous small variants, calculated as the number of phased SNVs divided by the number of heterozygous SNVs. |