Genomic Data Compression
Lossless genomic data compression
Technology from Enancio reduces genomic data storage and transfer costs

Benefits of Genomic Data Compression
Illumina is committed to delivering innovative sequencing technologies, and to helping customers manage growing volumes of next-generation sequencing (NGS) data output. Lossless genomic data compression technology from Enancio, formerly known as Lena and now known as original read archive (ORA) compression, offers optimal levels of speed and efficiency.
Genomic data compression allows for:
- Lower data storage costs
- High-speed data file transfers
- Reduced internal network traffic
Lossless Genomic Data Compression Technology
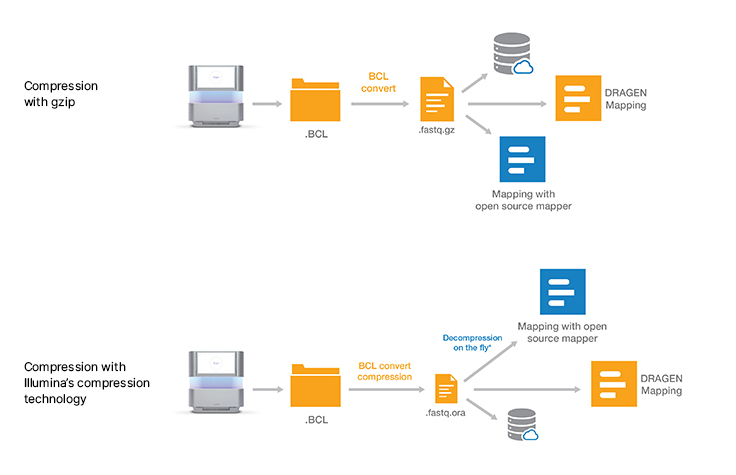
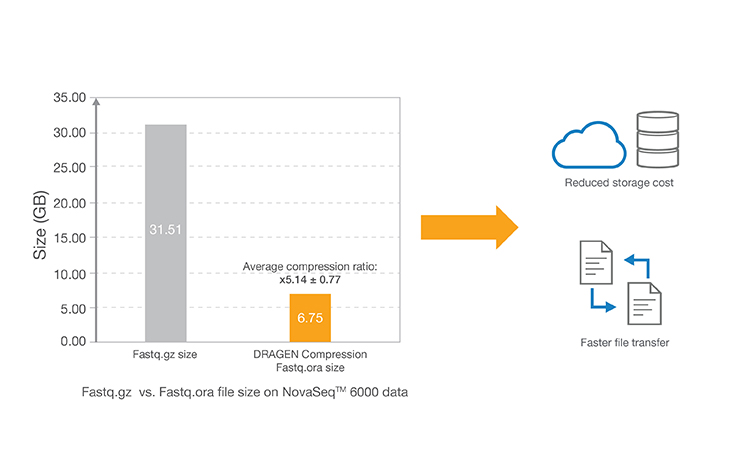
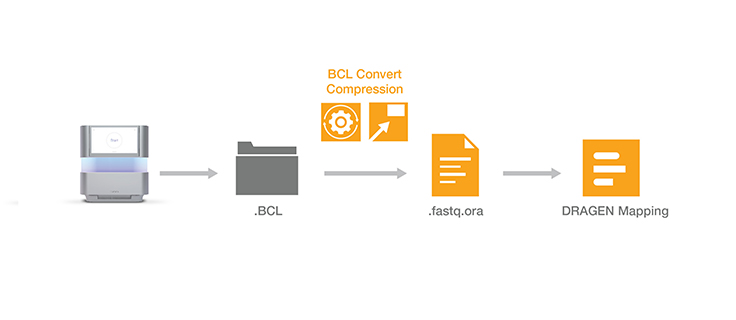
Lossless genomic data compression technology reduces the data storage footprint by as much as five times by compressing the output from Illumina sequencing systems. ORA compression technology uses a reference-based compression method. The idea is to use an ultra-fast mapping scheme to map reads onto a reference genome, and then store only the data needed to regenerate those reads: a position and a list of differences.
Other data compression technologies usually suffer from low speed. ORA compression technology is optimized for high compression ratios, as well as fast compression and decompression rates, while preserving data integrity. Quality scores are encoded in a lossless way using a range encoder and context models adapted to the different types of quality schemes.
Access DRAGEN ORA Decompression Software
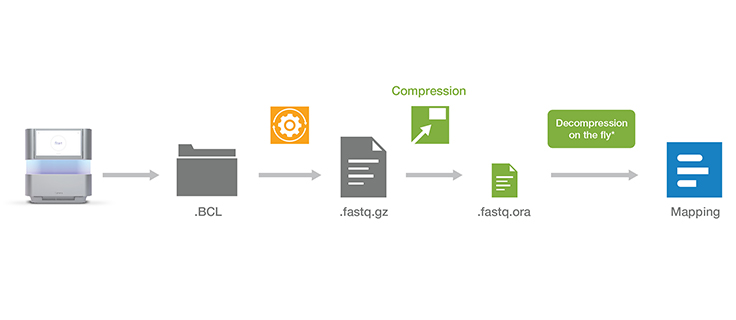
All files compressed with ORA compression technology can easily be decompressed using our decompression software. The decompression software is free to download and use.
Download decompression softwareOnce the decompression software is installed, a simple command can be used to directly pipe the output of decompression on the fly into a wide range of popular mapping tools such as BWA, STAR, and Bowtie. The compression and decompression technology is also integrated within DRAGEN secondary analysis software, which provides accurate, ultra-rapid analysis of sequencing data.

Lossless genomic compression available on-instrument
DRAGEN ORA lossless genomic data compression is now available on-instrument with the NextSeq 1000 and NextSeq 2000 Systems and NovaSeq X Series as well as on the DRAGEN secondary analysis server starting with v3.8. Learn more about:
NextSeq 1000/2000 Systems
NovaSeq X Series
DRAGEN secondary analysis
Compression Technology FAQs
Related Solutions
Genomic Data Storage & Security
Securely store, process, and share large genomic and NGS datasets in the cloud with built-in speed and scalability.
Sequencing Data Analysis
Our sequencing data analysis software helps you spend more time doing research, and less time configuring and running analysis workflows.
Illumina Informatics Product Portfolio
Explore a broad range of informatics products designed to simplify genomic data analysis and management.
Have questions about compression technology?
Contact us to learn more.
References
- On files generated by the NextSeq 1000 and NextSeq 2000 Systems and NovaSeq 6000 System.
- This result was obtained from the DNA sample NA12878 sequenced on the NovaSeq 6000 System with 30x coverage. Data is accessible in this BaseSpace project: basespace.illumina.com/s/3ExEZMlH8Lkq.
- Li H. and Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009 Jul 15; 25(14): 1754–1760.
- Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan; 29(1): 15–21.
- Langmead B. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 2009 10:R25