Introduction
Millions of human genomes and exomes have been sequenced, but their clinical applications remain limited due to the difficulty of distinguishing disease-causing mutations from benign genetic variation1,2. Because of their deleterious effects on fitness, clinically significant genetic variants tend to be extremely rare in the population3. Therefore, the observation of a variant at high frequencies in the population is strong evidence in favor of benign consequence2,4, enabling pathogenic mutations to be systematically identified by process of elimination. Assaying common variation across diverse human populations is an effective strategy for cataloguing benign variants5, but the total amount of common variation in present day humans is limited. Out of more than 70 million potential missense variants in the reference genome, only roughly 1 in 1000 are present at greater than 0.1% overall population allele frequency5,6.
Outside of modern human populations, chimpanzees comprise the next closest extant species, and share 99.4% amino acid sequence identity7. The near-identity of protein-coding sequence in humans and chimpanzees suggests that natural selection operating on chimpanzee protein-coding variants might also model the consequences on fitness of human identical mutations. If polymorphisms that are identical-by-state similarly affect fitness in the two species, the presence of a variant at high allele frequencies in chimpanzee populations should indicate benign consequence in human, expanding the catalog of known benign variants substantially. This formulates the hypothesis that needs to be verified with chimpanzee variants.
We demonstrated that common primate variants tend to be benign in human population. Using hundreds of thousands of common variants from population sequencing of six non-human primate species as training data, we developed PrimateAI, a deep neural network that predicts pathogenic mutations with high accuracy.
Common variants in other primates are largely benign in human
The recent availability of aggregated exome data, comprising 123,136 humans collected in the Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD), allows us to measure the impact of natural selection on missense and synonymous mutations across the allele frequency spectrum5. Singleton variants (observed only once in the cohort) closely match the expected 2.2:1 missense:synonymous ratio predicted by de novo mutation after adjusting for confounding factors (Fig. 1a)8, but at higher allele frequencies the number of observed missense variants decreases due to the purging of deleterious mutations by natural selection.
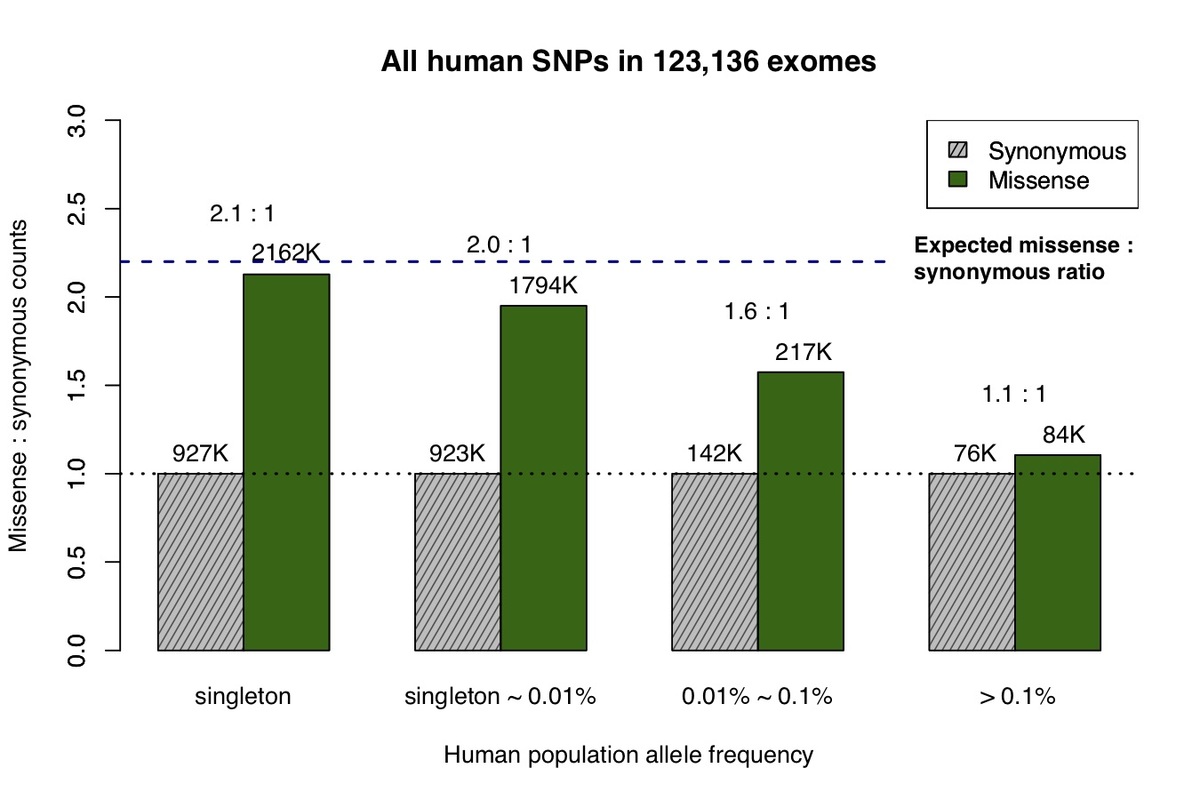
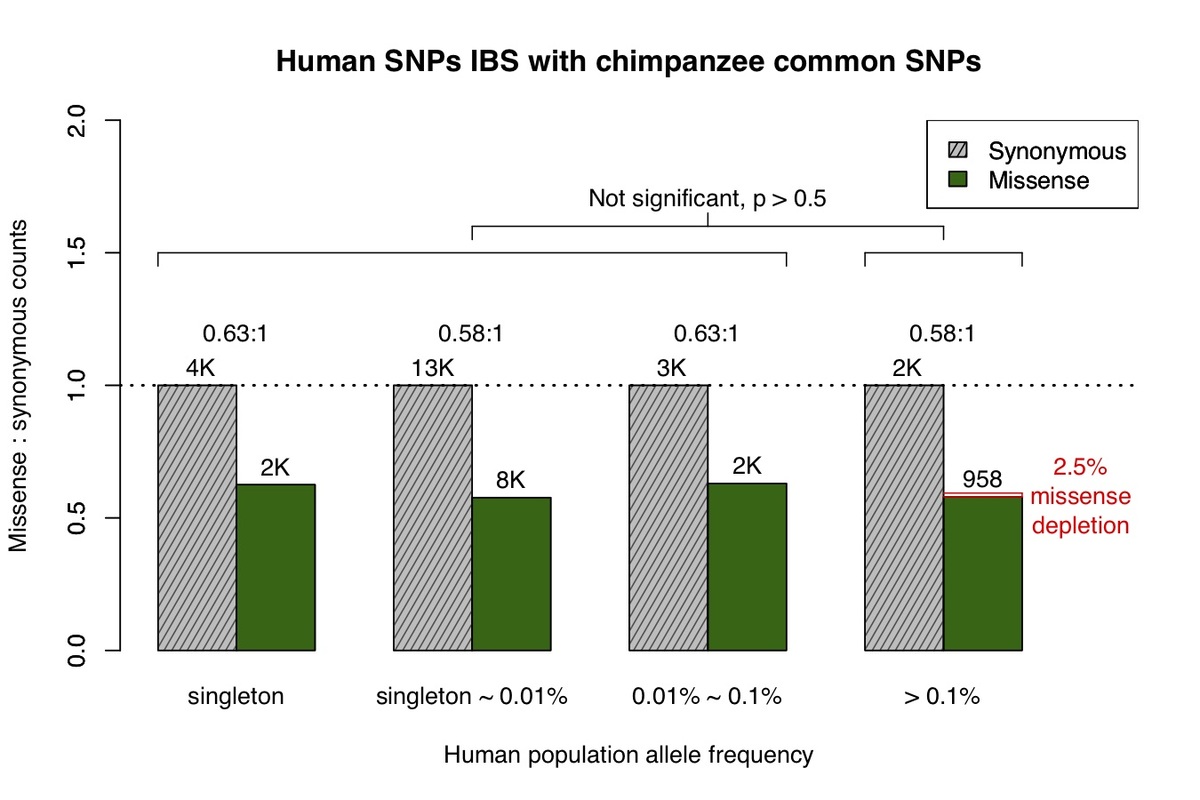
a, All missense and synonymous variants observed in the ExAC / gnomAD database were divided into 4 categories by allele frequency. Shaded grey bars represent counts of synonymous variants in each category; dark green bars represent missense variants. The height of each bar is scaled to the number of synonymous variants in each allele frequency category. b, Allele frequency spectrum for human missense and synonymous variants that are identical-by-state (IBS) with chimpanzee common variants.
Primate variants were obtained from the great ape genome sequencing project and dbSNP9,10. We first examined common chimpanzee variants that are identical-by-state with human variants (Fig. 1b), and discovered the missense:synonymous ratio is largely constant across the human allele frequency spectrum, which is consistent with absence of negative selection against common chimpanzee variants in the human population. The low missense:synonymous ratio observed in human variants that are identical-by-state with common chimpanzee variants is consistent with the larger effective population size in chimpanzee, which enables more efficient filtering of mildly deleterious variation11,12.
We next identified human variants that are identical-by-state with variation observed in at least one of six non-human primate species. Variation in each of the six species largely represent common variants based on the limited number of individuals sequenced and the low missense:synonymous ratios observed for each species. Similar to chimpanzee, we find that the missense:synonymous ratios for variants from the six non-human primate species are roughly equal across the human allele frequency spectrum, other than a mild depletion of missense variation at common allele frequencies (Fig. 2), which is expected due to the inclusion of a minority of rare variants.
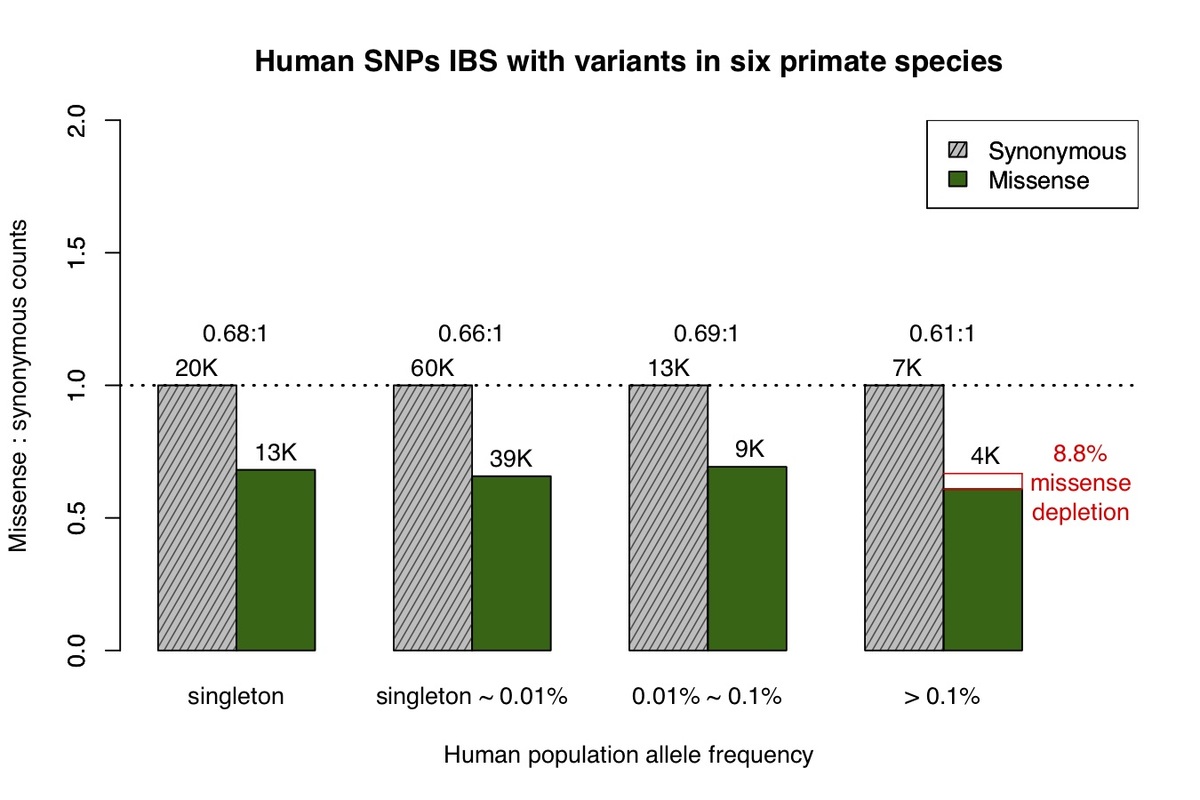
Figure 2. Allele frequency spectrum for human missense and synonymous variants that are observed in at least one of the non-human primate species, similar to Fig.1.
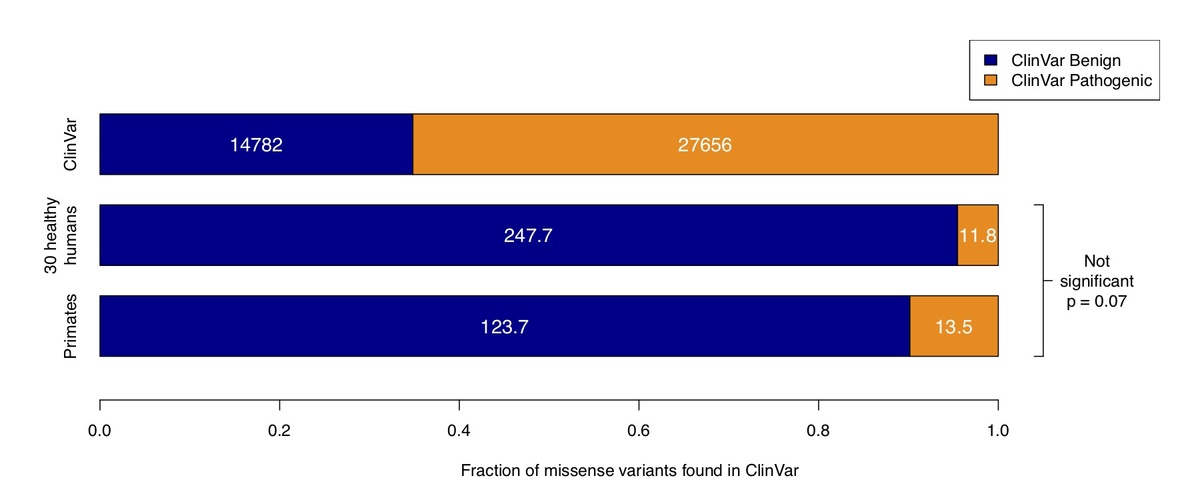
Figure 3. Counts of benign and pathogenic missense variants in the overall ClinVar database (top row), compared to ClinVar variants in a cohort of 30 humans simulated by sampling from ExAC / gnomAD allele frequencies (middle row), compared to variants observed in primates (bottom row). Conflicting benign and pathogenic assertions and variants annotated only with uncertain significance were excluded.
A deep learning network for variant pathogenicity classification
The importance of variant classification for clinical applications has inspired numerous attempts to use supervised machine learning to address the problem, but these efforts have been hindered by the lack of an adequately-sized truth dataset containing confidently labeled benign and pathogenic variants for training14-24. Existing databases of human expert curated variants cover a small fraction of the genome, with ~50% of the variants in the ClinVar database coming from only 200 genes (~1% of human protein-coding genes). Moreover, systematic studies reveal that many human expert annotations have questionable supporting evidence5,25, underscoring the difficulty of interpreting rare variants that may be observed in only a single patient. To reduce human interpretation biases, recent classifiers have been trained on common human polymorphisms or fixed human-chimpanzee substitutions26-29, but these classifiers also use as their input the prediction scores of earlier classifiers that were trained on human curated databases. Objective benchmarking of the performance of these various methods has been elusive in the absence of an independent, bias-free truth dataset30.
Variation from the six non-human primates (chimpanzee, bonobo, gorilla, orangutan, rhesus, and marmoset) contributes over 300,000 unique missense variants that are non-overlapping with common human variation, and largely represent common variants of benign consequence that have been through the sieve of purifying selection, greatly enlarging the training dataset available for machine learning approaches. On average, each primate species contributes the equivalent of 50K variants, more variants than the current total in the whole of the ClinVar database. Additionally, this content is free from biases in human interpretation.
Using a dataset consisting of common human variants and primate variation, we trained a novel deep residual network, PrimateAI (https://github.com/Illumina/PrimateAI), which takes as input the amino acid sequence flanking the variant of interest and the orthologous sequence alignments in other species (Fig. 4a)31. Unlike existing classifiers which employ human-engineered features, our deep learning network learns to extract features directly from primary sequence. To incorporate information about protein structure, we trained separate networks to predict secondary structure and solvent accessibility from sequence alone32,33, and then included these as sub-networks in the full model (Fig. 4b). Given the small number of human proteins that have been successfully crystallized, inferring structure from primary sequence has the advantage of avoiding biases due to incomplete protein structure and functional domain annotation. The total depth of the network, with protein structure included, was 36 layers of convolutions, consisting of roughly 400,000 trainable parameters.
To train a classifier using only variants with benign labels, we framed the prediction problem as whether a given mutation is likely to be observed as a common variant in the population. Several factors influence the probability of observing a variant at high allele frequencies, of which we are interested only in deleteriousness. We matched each variant in the benign training set with a unlabeled missense mutation, controlling for the confounding factors, and trained the deep learning network to distinguish between benign variants and matched controls8. As the number of unlabeled variants greatly exceeds the size of the labeled benign training dataset, we trained eight networks in parallel, each using a different set of unlabeled variants matched to the benign training dataset, to obtain a consensus prediction.
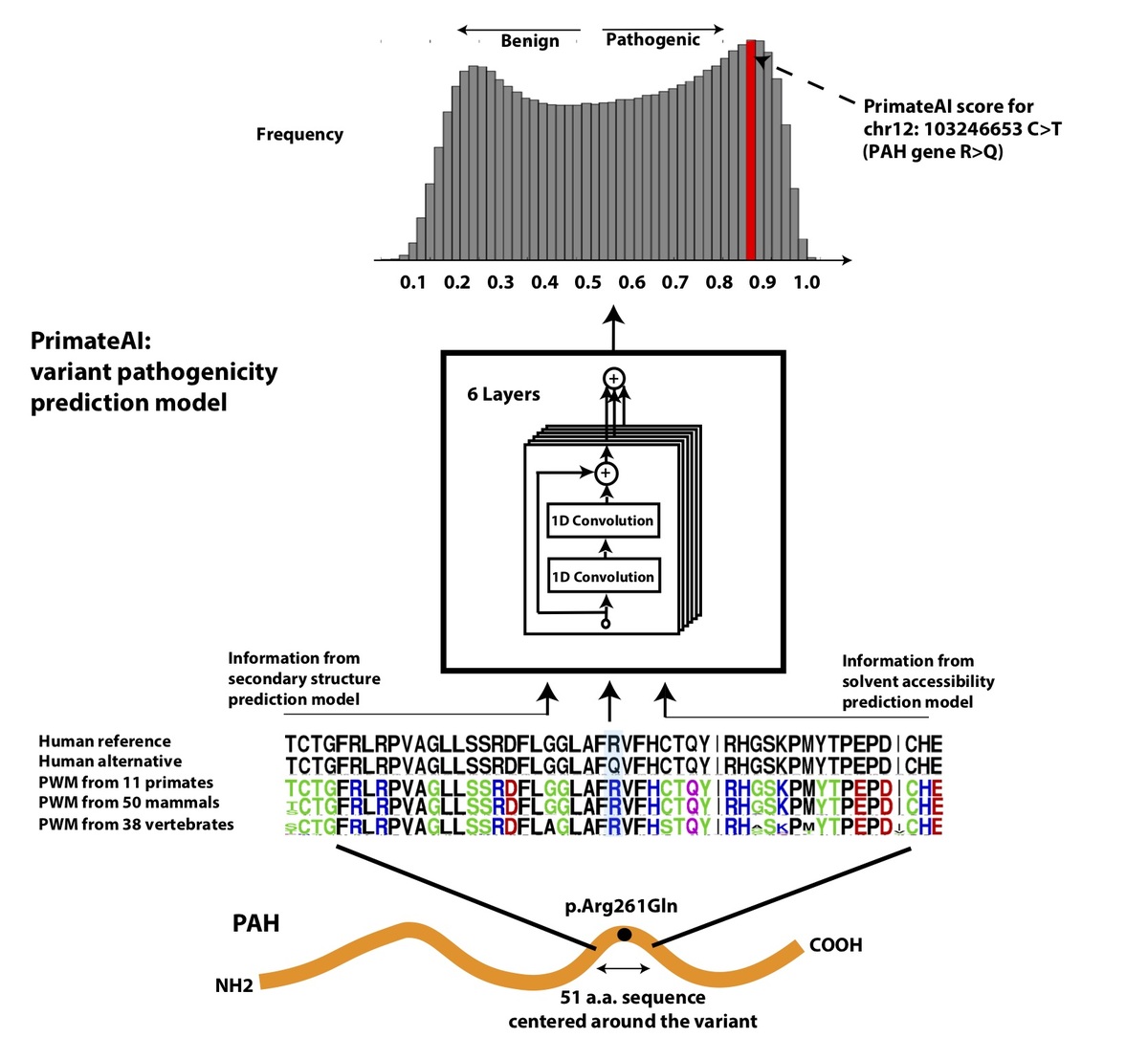
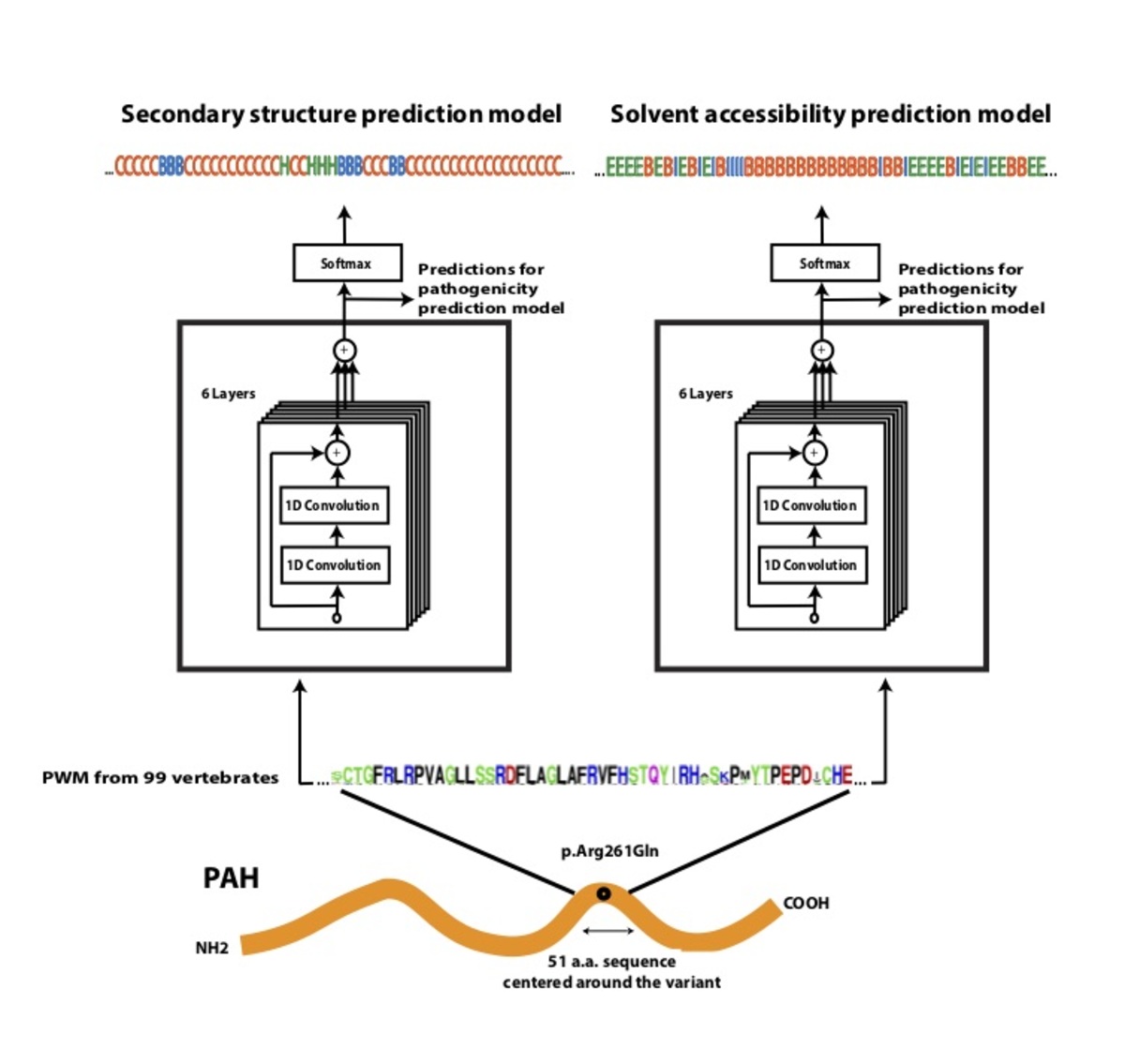
Figure 4. Architecture of PrimateAI, the deep learning network for pathogenicity prediction. a, Architecture of the deep learning network for pathogenicity prediction, PrimateAI. Predicted pathogenicity, denoted as PrimateAI score, is on a scale from 0 (benign) to 1 (pathogenic). The network takes as input the human amino acid (AA) reference and alternate sequence (51 AAs) centered at the variant, the position weight matrix (PWM) conservation profiles calculated from 99 vertebrate species, and b, the outputs of secondary structure and solvent accessibility prediction deep learning networks, which predict three-state protein secondary structure (helix—H, beta sheet—B, and coil—C) and three-state solvent accessibility (buried—B, intermediate—I, and exposed—E).
Example of Pathogenicity Prediction
Using only primary amino acid sequence as its input, the deep learning network accurately assigns high pathogenicity scores to residues at critical protein functional domains, as shown for the voltage-gated sodium channel SCN2A (Fig. 5), a major disease gene in epilepsy, autism, and intellectual disability. The structure of the SCN2A consists of four homologous repeats, each containing six transmembrane helixes (S1-S6)34,35. Upon membrane depolarization, the positively-charged S4 transmembrane helix moves towards the extracellular side of the membrane, causing the S5/S6 pore-forming domains to open via the S4-S5 linker. Mutations in the S4, S4-S5 linker, and S5 domains, which are clinically associated with early onset epileptic encephalopathy36, are predicted by the network to have the highest pathogenicity scores in the gene, and are depleted for variants in the healthy population.
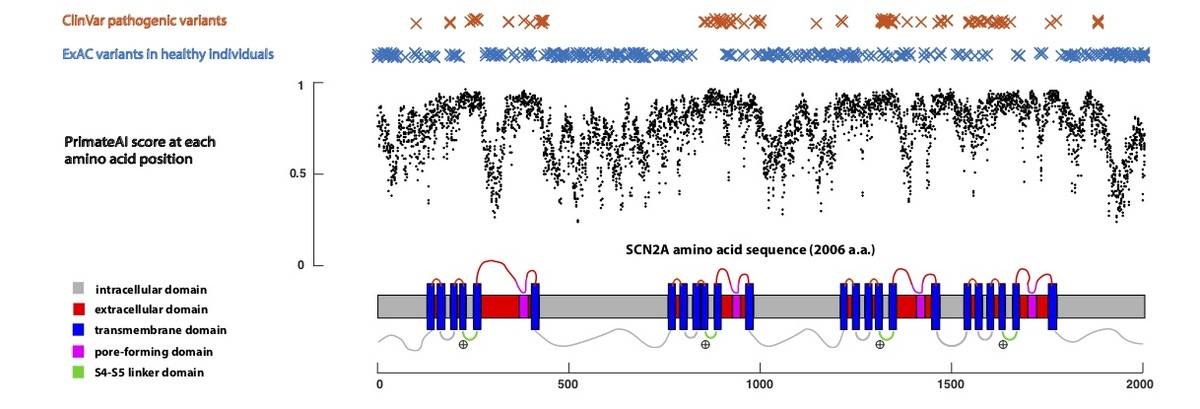
Figure 5. Predicted pathogenicity score at each amino acid position in the SCN2A gene, annotated for key functional domains. Plotted along the gene is the average PrimateAI score for missense substitutions at each amino acid position.
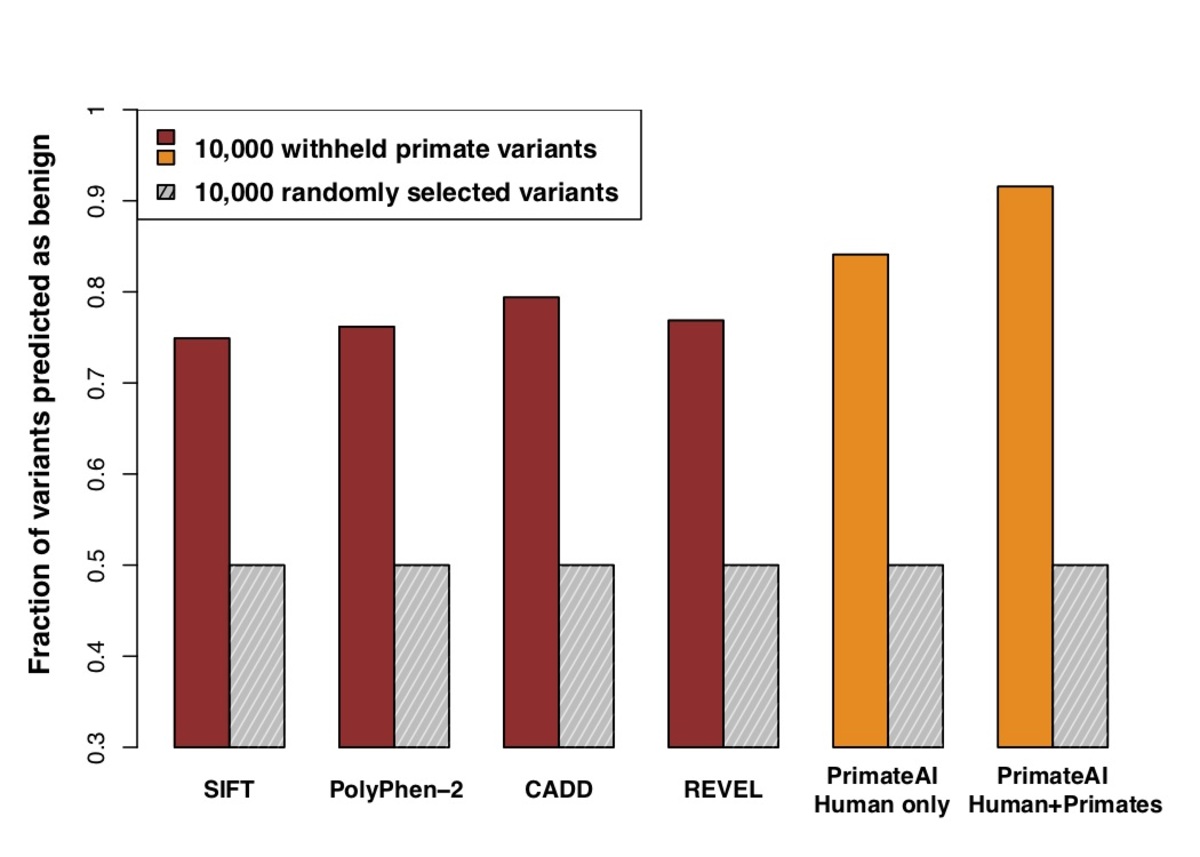
Figure 6. Comparison of classifiers at predicting benign consequence for a test set of 10,000 common primate variants that were withheld from training. The y-axis represents the percentage of primate variants correctly classified as benign, after normalizing the threshold of each classifier to its 50th percentile score on a set of 10,000 random variants that were matched for mutational rate.
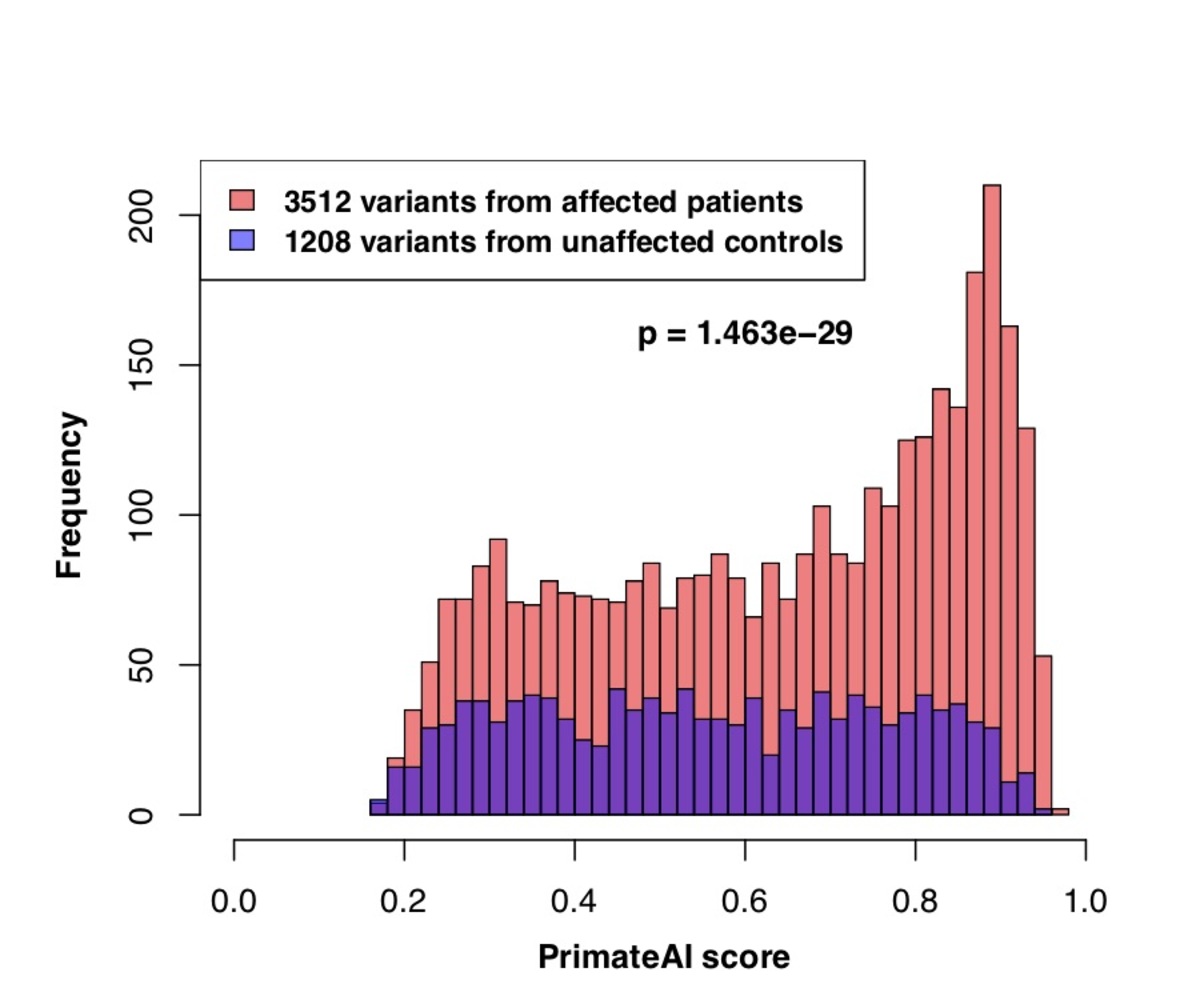
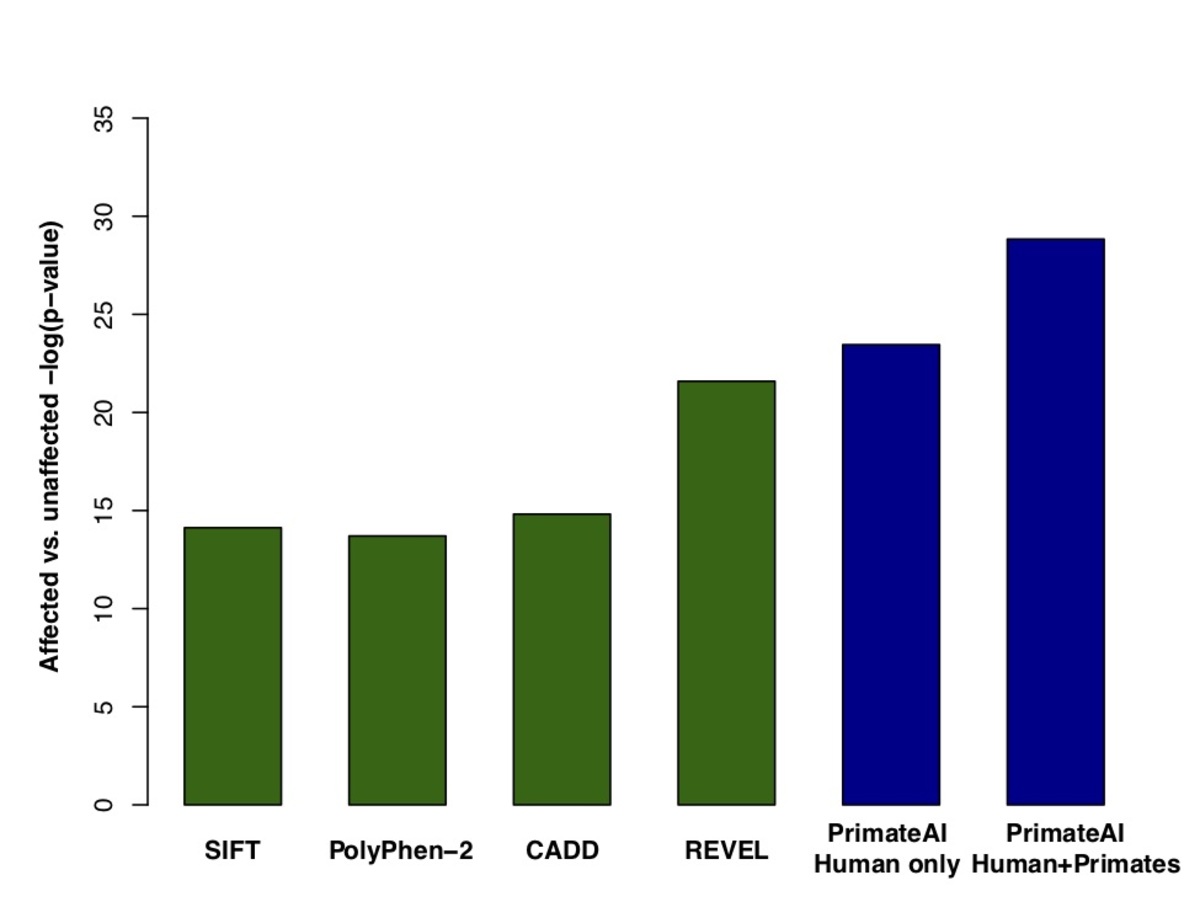
Figure 7. a, Distributions of PrimateAI prediction scores for de novo missense variants occurring in DDD patients compared to unaffected siblings, with corresponding Wilcoxon rank-sum p-value. b, Comparison of classifiers at separating de novo missense variants in DDD cases versus controls. Wilcoxon rank-sum test p-values are shown for each classifier.
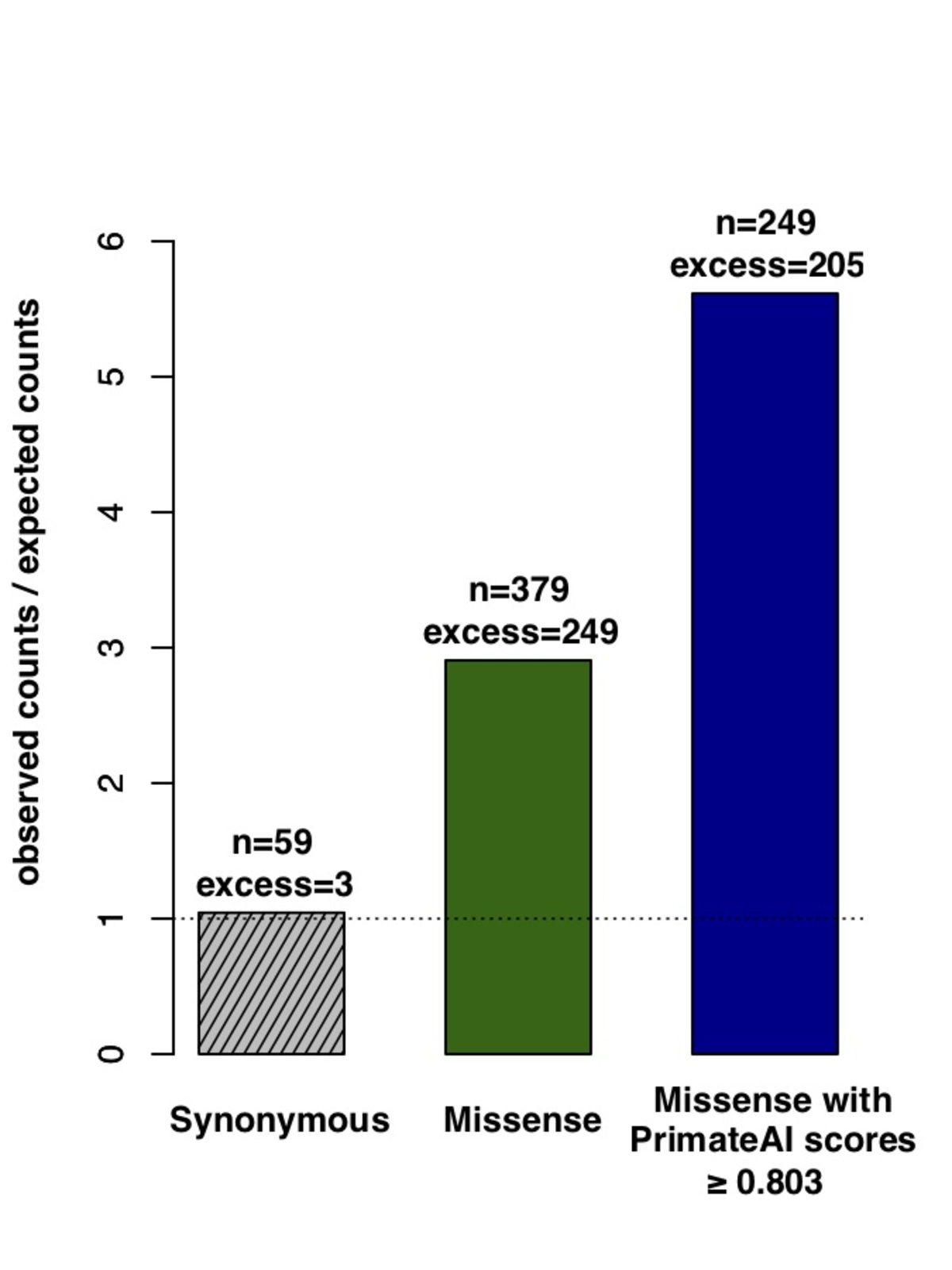
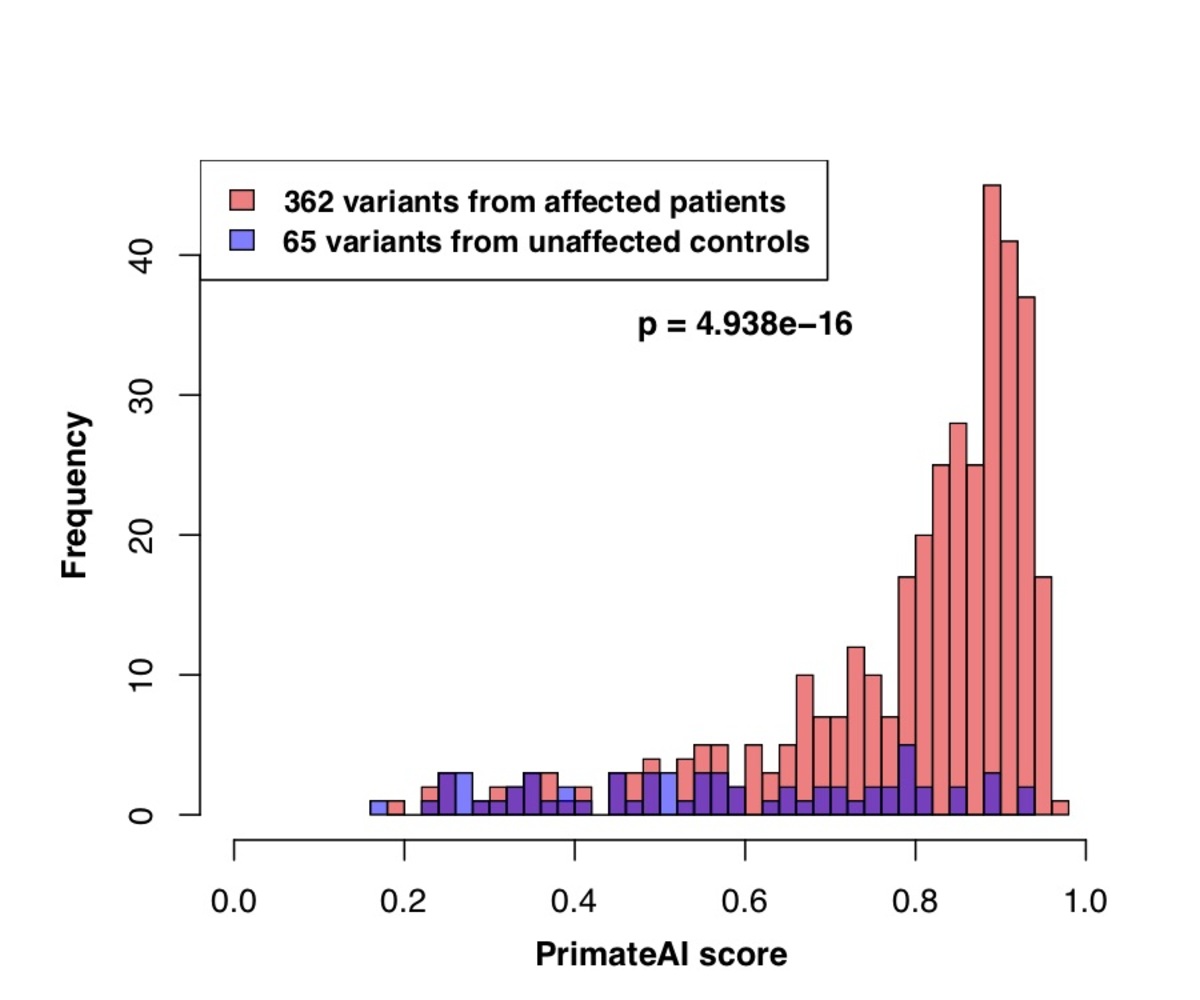
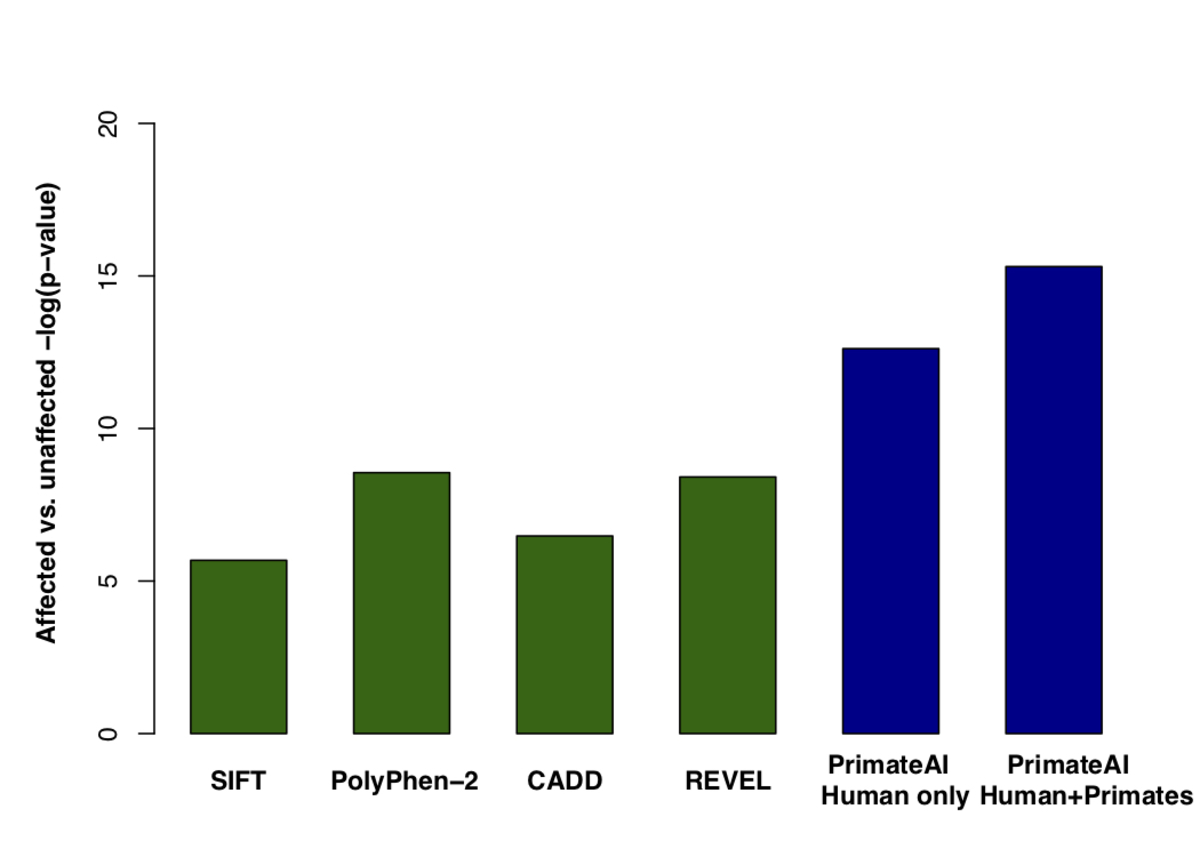
Figure 8. Classification accuracy within 605 DDD genes with P < 0.05. a, Enrichment of de novo missense mutations over expectation in affected individuals from the DDD cohort within 605 associated genes that were significant for de novo protein truncating variation (p<0.05). b, Distributions of PrimateAI prediction scores for de novo missense variants occurring in DDD patients vs unaffected siblings within the 605 associated genes, with corresponding Wilcoxon rank-sum p-value. c, Comparison of various classifiers at separating de novo missense variants in cases vs controls within the 605 genes. The y-axis shows the p-values of the Wilcoxon rank-sum test for each classifier.
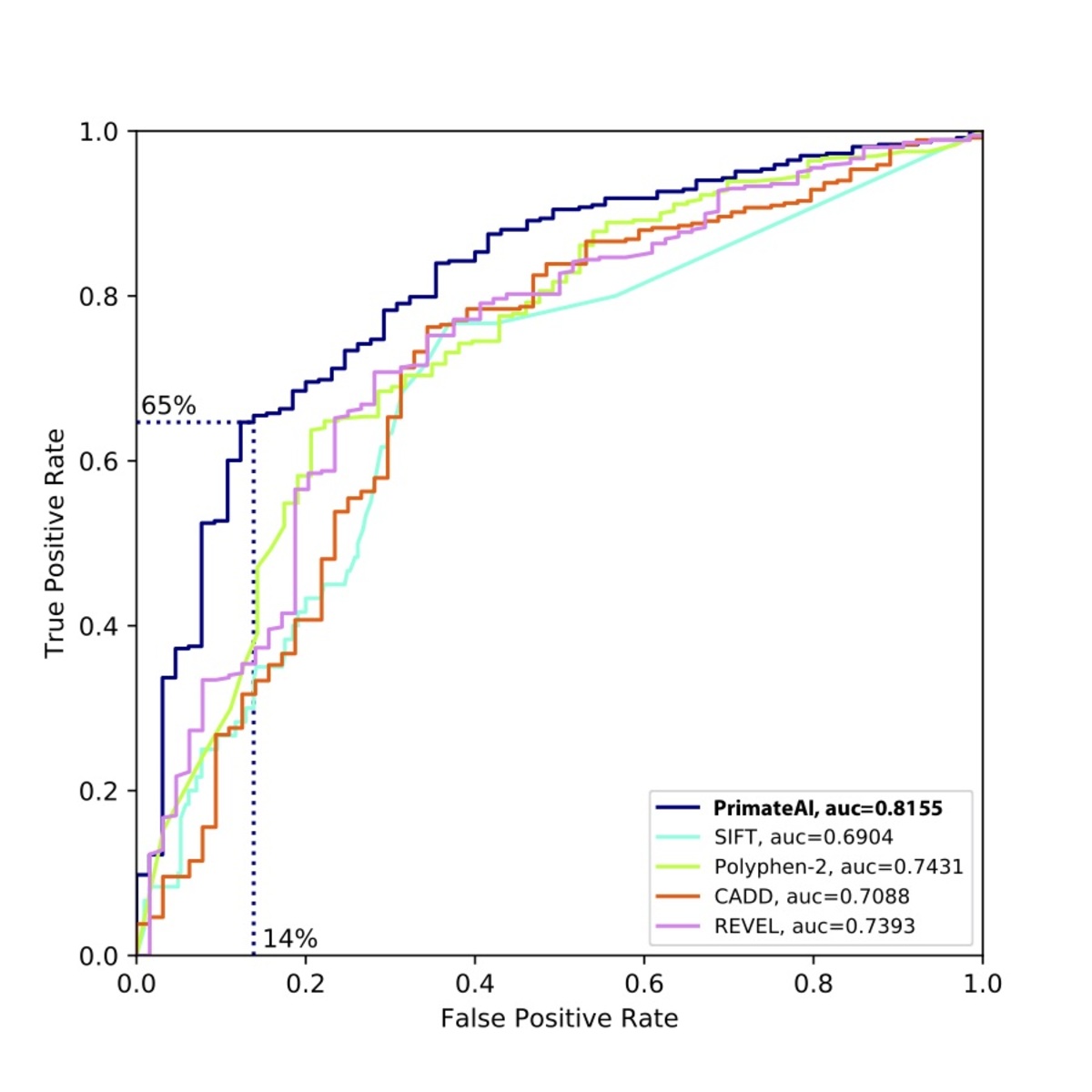
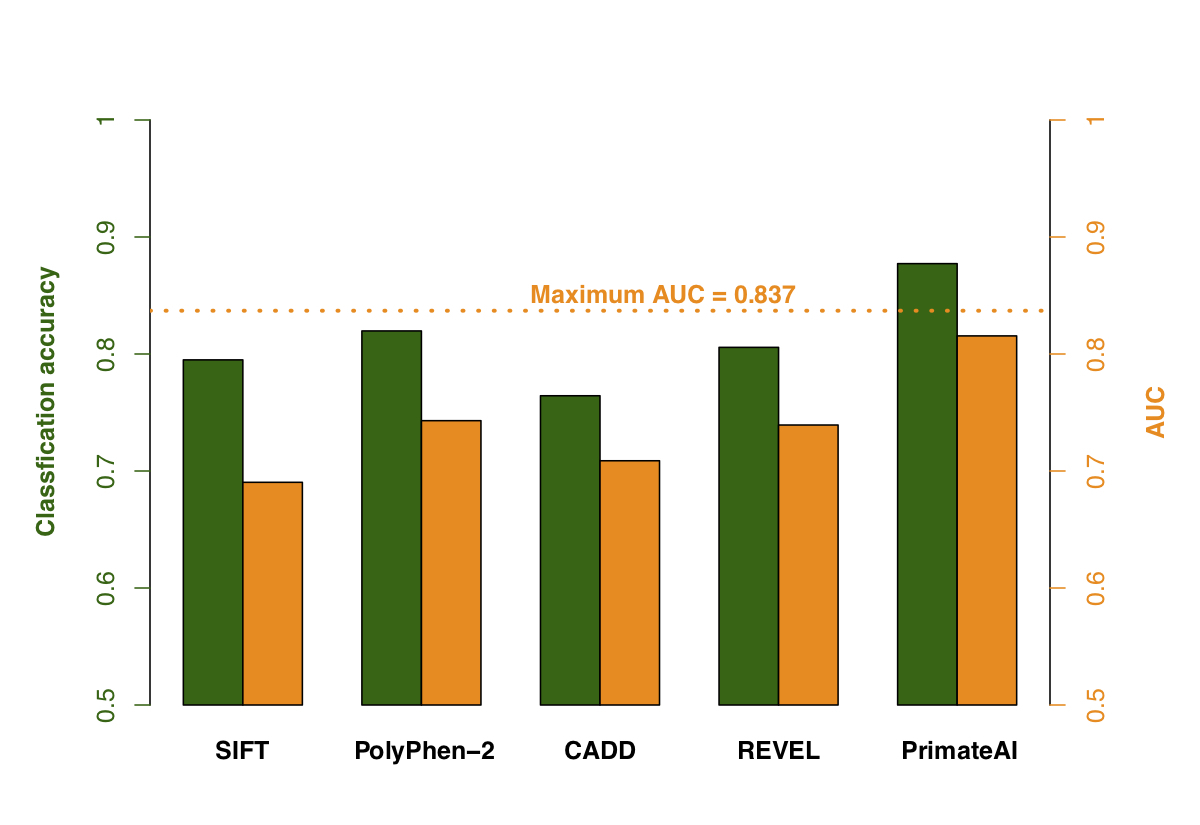
Figure 9. Comparison of various classifiers. a, Performance shown on a Receiver Operator Characteristic (ROC) curve, with area under the curve (AUC) indicated for each classifier. b, Classification accuracy and AUC for each classifier. The classification accuracy shown is the average of the true positive and true negative error rates, using the threshold where the classifier would predict the same number of pathogenic and benign variants as expected based on the enrichment in Fig. 8a. To take into account that 33% of the DDD de novo missense variants represent background, the maximum achievable AUC for a perfect classifier is indicated with a dotted line.
Our results suggest that systematic primate population sequencing is an effective strategy to classify the millions of human variants of uncertain significance that currently limit clinical genome interpretation. The accuracy of our deep learning network on both withheld common primate variants and clinical variants increases with the number of benign variants used to train the network. Cataloging common variation from additional primate species would improve interpretation for millions of variants of uncertain significance, further advancing the clinical utility of human genome sequencing.
Acknowledgements
We would like to thank J. K. Pritchard, M. E. Hurles, J. W. Belmont, and R. E. Green for insightful discussions. We would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. Yanjun Li and Xiaolin Li were partially supported by R01GM110240 from the National Institute of General Medical Sciences and National Science Foundation (grants CNS- 1747783, CNS- 1624782, and OAC-1229576). We would like to acknowledge the authors in the original paper, including Laksshman Sundaram, Samskruthi Reddy Padigepati, Jeremy F. McRae, Yanjun Li, Jack A. Kosmicki, Nondas Fritzilas, Jorg Hakenberg, Anindita Dutta, John Shon, Jinbo Xu, Serafim Batzloglou, and Xiaolin Li.
External links
Publication: https://pubmed.ncbi.nlm.nih.gov/30038395/
Software: https://github.com/Illumina/PrimateAI
Primate polymorphisms from the great ape genome project:
https://eichlerlab.gs.washington.edu/greatape/data.html
And from dbSNP database: https://www.ncbi.nlm.nih.gov/snp/
PrimateAI scores of 70 million variants: https://basespace.illumina.com/s/cPgCSmecvhb4
References
- MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469-476, doi:10.1038/nature13127 (2014).
- Rehm, H. L., J. S. Berg, L. D. Brooks, C. D. Bustamante, J. P. Evans, M. J. Landrum, D. H. Ledbetter, D. R. Maglott, C. L. Martin, R. L. Nussbaum, S. E. Plon, E. M. Ramos, S. T. Sherry, M. S. Watson. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 372, 2235-2242 (2015).
- Bamshad, M. J., S. B. Ng, A. W. Bigham, H. K. Tabor, M. J. Emond, D. A. Nickerson, J. Shendure. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755 (2011).
- Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405-424, doi:10.1038/gim.2015.30 (2015).
- Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291, doi:10.1038/nature19057 (2016).
- Liu, X., X. Jian, E. Boerwinkle. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. . Human Mutation 32, 894–899 (2011).
- Chimpanzee Sequencing Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69-87, doi:10.1038/nature04072 (2005).
- Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944-950, doi:10.1038/ng.3050 (2014).
- Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308-311, doi:10.1093/nar/29.1.308 (2001).
- Prado-Martinez, J. et al. Great ape genome diversity and population history. Nature 499, 471-475 (2013).
- Kimura, M. The neutral theory of molecular evolution. Cambridge University Press, 1983
- de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477-481, doi:10.1126/science.aag2602 (2016).
- Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862-868, doi:10.1093/nar/gkv1222 (2016).
- Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res 11, 863-874, doi:10.1101/gr.176601 (2001).
- Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248-249, doi:10.1038/nmeth0410-248 (2010).
- Chun, S., J. C. Fay. Identification of deleterious mutations within three human genomes. Genome Research 19, 1553-1561 (2009).
- Schwarz, J. M., C. Rödelsperger, M. Schuelke, D. Seelow. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
- Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39, e118, doi:10.1093/nar/gkr407 (2011).
- Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24, 2125-2137, doi:10.1093/hmg/ddu733 (2015).
- Carter, H., Douville, C., Stenson, P. D., Cooper, D. N. & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 Suppl 3, S3, doi:10.1186/1471-2164-14-S3-S3 (2013).
- Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
- Gulko, B., Hubisz, M. J., Gronau, I. & Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet 47, 276-283, doi:10.1038/ng.3196 (2015).
- Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536-1543, doi:10.1093/bioinformatics/btv009 (2015).
- Quang, D., Chen, Y. & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761-763, doi:10.1093/bioinformatics/btu703 (2015).
- Bell, C. J., D. L. Dinwiddie, N. A. Miller, S. L. Hateley, E. E. Ganusova, J. Midge, R. J. Langley, L. Zhang, C. L. Lee, R. D. Schilkey, J. E. Woodward, H. E. Peckham, G. P. Schroth, R. W. Kim, S. F. Kingsmore. Comprehensive carrier testing for severe childhood recessive diseases by next generation sequencing. Sci. Transl. Med. 3, 65ra64 (2011).
- Kircher, M., D. M. Witten, P. Jain, B. J. O’Roak, G. M. Cooper, J. Shendure. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310-315 (2014).
- Smedley, D. et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet 99, 595-606, doi:10.1016/j.ajhg.2016.07.005 (2016).
- Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99, 877-885, doi:10.1016/j.ajhg.2016.08.016 (2016).
- Jagadeesh, K. A., A. M. Wenger, M. J. Berger, H. Guturu, P. D. Stenson, D. N. Cooper, J. A. Bernstein, G. Bejerano. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nature Genetics 48, 1581-1586 (2016).
- Grimm, D. G. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human Mutation 36, 513-523 (2015).
- He, K., X. Zhang, S. Ren, J. Sun. Proceedings of the IEEE conference on computer vision and pattern recognition. IEEE 770-778.
- Heffernan, R. et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci Rep 5, 11476, doi:10.1038/srep11476 (2015).
- Wang, S., J. Peng, J. Ma, J. Xu. Protein secondary structure prediction using deep convolutional neural fields. Scientific Reports 6, 18962-18962 (2016).
- Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. https://www.nature.com/articles/nature10238
- Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. https://science.sciencemag.org/content/355/6328/eaal4326
- Nakamura, K. et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81, 992-998, doi:10.1212/WNL.0b013e3182a43e57 (2013).
- Vissers, L. E., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17, 9-18, doi:10.1038/nrg3999 (2016).
- Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242-245, doi:10.1038/nature11011 (2012).
- Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237-241, doi:10.1038/nature10945 (2012).
- De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209-215, doi:10.1038/nature13772 (2014).
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223-228, doi:10.1038/nature14135 (2015).
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433-438, doi:10.1038/nature21062 (2017).
- Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216-221, doi:10.1038/nature13908 (2014).
- Zhu, X., Need, A. C., Petrovski, S. & Goldstein, D. B. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 17, 773-781, doi:10.1038/nn.3713 (2014).