Exome sequencing has transformed the clinical diagnosis of patients and families with rare genetic disorders and, when employed as a first-line test, significantly reduces the time and costs of the diagnostic odyssey. However, the diagnostic yield of exome sequencing is ~25-30% in rare genetic disease cohorts1, leaving many patients without a diagnosis. Whole genome sequencing could substantially increase the diagnostic yield if we have methods that can identify functional noncoding variants.
Cryptic splice variants, which are variants that disrupt the normal pattern of mRNA splicing despite lying outside the essential GT and AG splice dinucleotides, are a class of noncoding variants that have long been recognized to play a significant role in rare genetic diseases. However, these mutations are often overlooked in clinical practice due to our incomplete understanding of the splicing code and the resulting difficulty in accurately identifying splice-altering variants.
Recently, RNA-seq has emerged as a promising assay for detecting such variants2, but thus far the utility of RNA-seq in a clinical setting remains limited to a minority of cases where the relevant cell type is accessible to biopsy. General prediction of splicing from arbitrary pre-mRNA sequence would potentially allow precise prediction of the splice-altering consequences of noncoding variants identified via whole genome sequencing, which could substantially improve diagnosis in patients with genetic diseases.
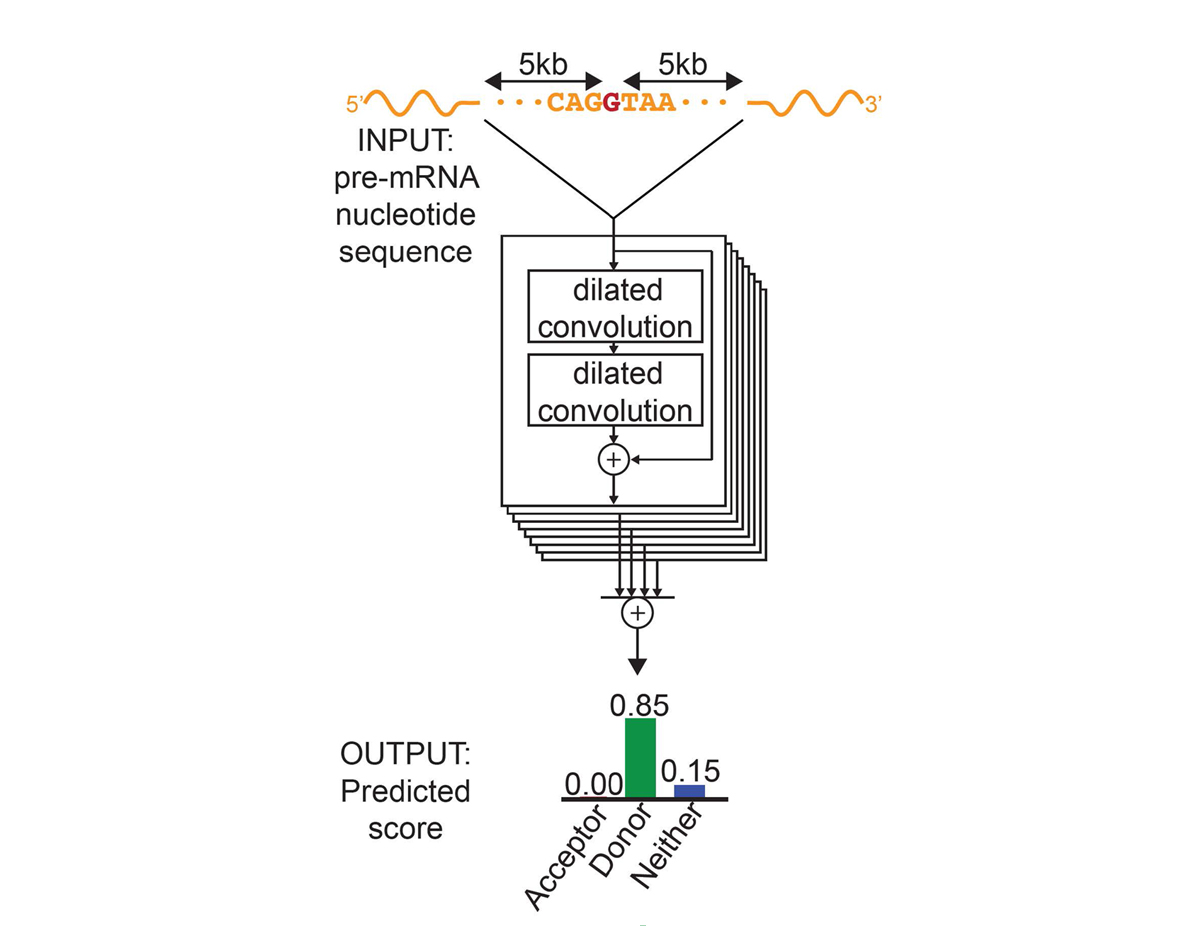
For each position in the pre-mRNA transcript, SpliceAI-10k uses 10,000 nucleotides of flanking sequence as input and predicts whether that position is a splice acceptor, splice donor, or neither.
We constructed SpliceAI, a deep residual neural network that predicts whether each position in a pre-mRNA transcript is a splice donor or splice acceptor based on the genomic sequence of the pre-mRNA transcript (Figure 1). Because splice donors and splice acceptors may be separated by thousands of nucleotides, we employed a neural network architecture that can recognize sequence determinants spanning very large genomic distances. In contrast to previous methods that have only considered short nucleotide windows adjoining exon-intron boundaries3, or relied on human-engineered features4, our neural network learns splicing determinants directly from the primary sequence by evaluating 10,000 nucleotides of flanking context sequence to predict the splice function of each position in the pre-mRNA transcript. We used GENCODE-annotated pre-mRNA transcript sequences on a subset of the human chromosomes to train the parameters of the neural network, and transcripts on the remaining chromosomes, with paralogs excluded, to test the network’s predictions.
For pre-mRNA transcripts in the test dataset, the network predicts splice junctions with 95% top-k accuracy, which is the percentage of correctly predicted splice sites at the threshold where the number of predicted sites is equal to the actual number of splice sites present in the test dataset (Figure 2). Even genes in excess of 100 kb such as CFTR are often reconstructed perfectly to nucleotide precision (Figure 3).
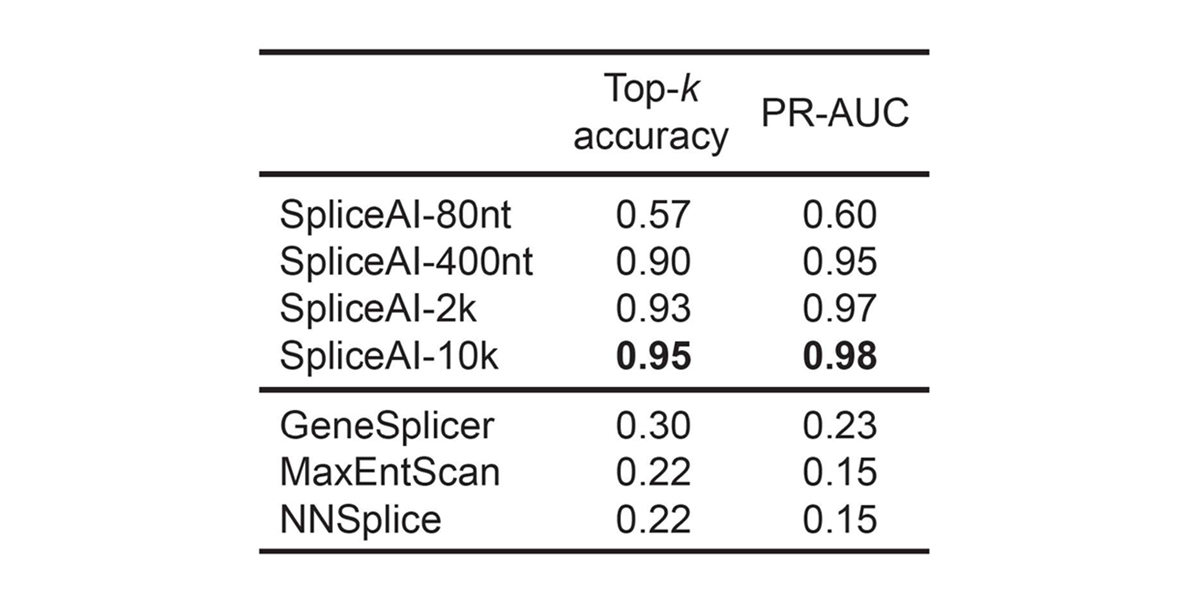
For pre-mRNA transcripts in the test dataset, the network predicts splice junctions with 95% top-k accuracy, which is the percentage of correctly predicted splice sites at the threshold where the number of predicted sites is equal to the actual number of splice sites present in the test dataset.
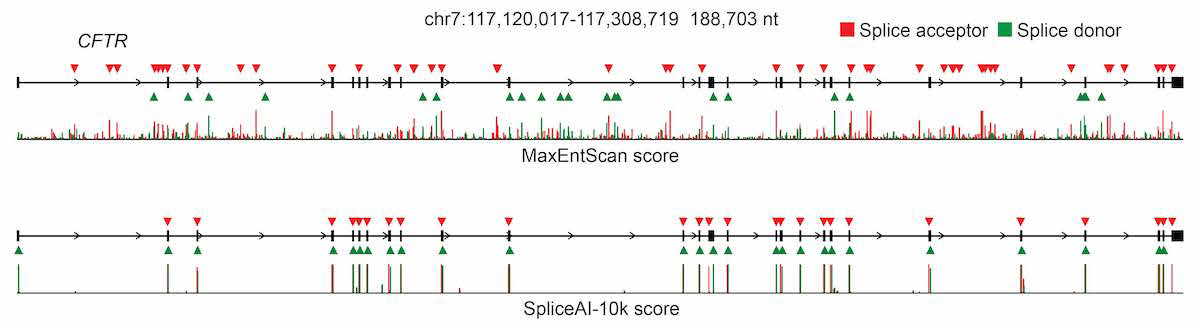
The full pre-mRNA transcript for the CFTR gene scored using MaxEntScan (top) and SpliceAI-10k (bottom) is shown, along with predicted acceptor (red arrows) and donor (green arrows) sites and the actual positions of the exons (black boxes). For each method, we applied the threshold that made the number of predicted sites equal to the total number of actual sites.
To confirm that the network is not simply relying on exonic sequence biases, we also tested the network on long noncoding RNAs. Despite the incompleteness of noncoding transcript annotations, the network predicts known splice junctions in lincRNAs with 84% top-k accuracy, indicating that it can approximate the behavior of the spliceosome on arbitrary sequences that are free from protein-coding selective pressures.
The network was only trained on reference transcript sequences and splice junction annotations, and variant data was not used during training, making prediction of variant effects a challenging test of the network’s ability to accurately model the sequence determinants of splicing. We extended the deep learning network to the evaluation of genetic variants for splice-altering function by predicting exon-intron boundaries for both the reference pre-mRNA transcript sequence and the alternate transcript sequence containing the variant, and taking the difference between the scores (referred to as ∆ Score, Figure 4).
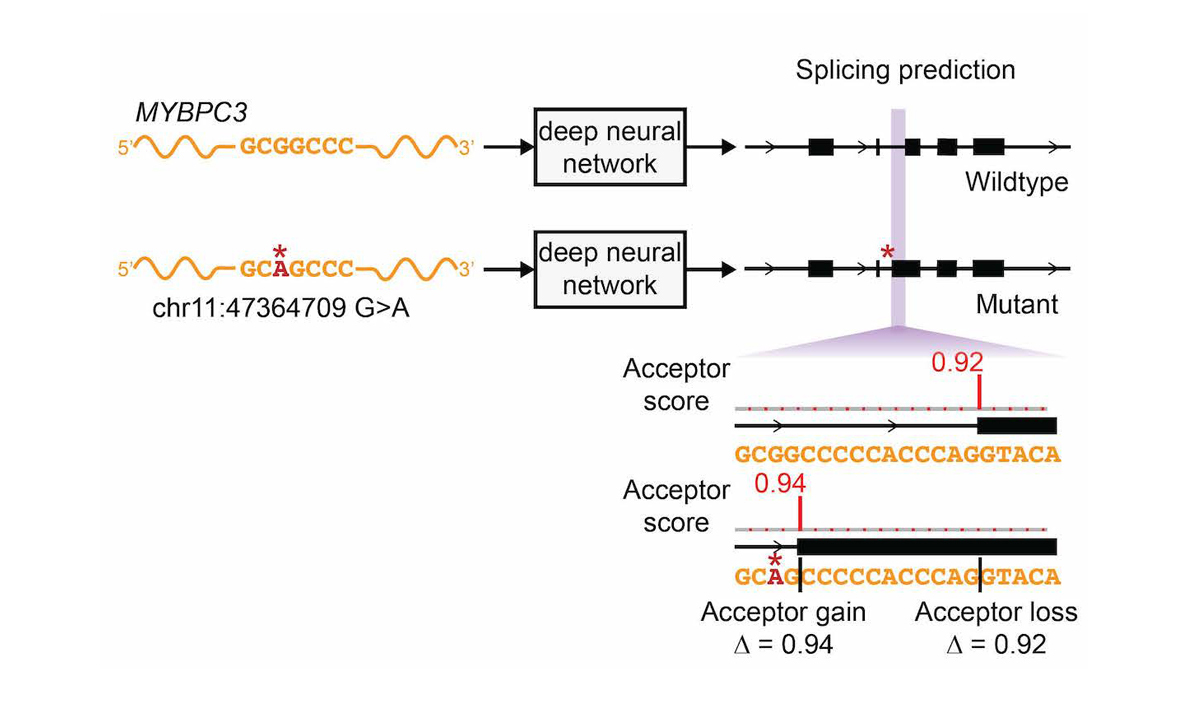
To assess the splice-altering impact of a mutation, SpliceAI-10k predicts acceptor and donor scores at each position in the pre-mRNA sequence of the gene with and without the mutation, as shown here for rs397515893, a pathogenic cryptic splice variant in the MYBPC3 intron associated with cardiomyopathy. The ∆ Score value for the mutation is the largest change in splice prediction scores within 50 nt from the variant.
We looked for the effects of cryptic splice variants in RNA-seq data in the GTEx cohort5, comprising 149 individuals with both whole genome sequencing and RNA-seq from multiple tissues. Confidently predicted cryptic splice variants (∆ Score > 0.5) validate on RNA-seq at three-quarters the rate of essential GT or AG splice disruptions (Figure 2D in SpliceAI paper). Both the validation rate and effect size of cryptic splice variants closely track their ∆ Scores, demonstrating that the model’s prediction score is a good proxy for the splice-altering potential of a variant. Validated variants, especially those with lower scores (∆ Score < 0.5), are often incompletely penetrant, and result in alternative splicing with production of a mixture of both aberrant and normal transcripts in the RNA-seq data. For cryptic splice variants that produce aberrant splice isoforms in at least three-tenths of the observed copies of the mRNA transcript, the network has a sensitivity of 71% when the variant is near exons, and 41% when the variant is in deep intronic sequence (∆ Score > 0.5, Figure 2F in SpliceAI paper). These findings indicate that deep intronic variants are more challenging to predict, possibly because deep intronic regions contain fewer of the specificity determinants that have been selected to be present near exons.
To explore the signature of natural selection on predicted cryptic splice variants, we scored each variant present in 60,706 human exomes from the Exome Aggregation Consortium (ExAC) database6 and in 15,496 humans from the Genome Aggregation Database (gnomAD) cohort7, and identified variants that were predicted to alter exon-intron boundaries. To measure the extent of negative selection acting on predicted splice-altering variants, we counted the number of predicted splice-altering variants found at common allele frequencies (> 0.1% in the cohort) and compared it to the number of predicted splice-altering variants at singleton allele frequencies. Because of the recent exponential expansion in human population size, singleton variants represent recently created mutations that have been minimally filtered by purifying selection. In contrast, common variants represent a subset of neutral mutations that have passed through the sieve of purifying selection. We observed that high-scoring cryptic splice variants are severely depleted in common variants indicating that these variants are under strong negative selection (Figures 4B and 4D in SpliceAI paper). This indicates that the vast majority of confidently predicted cryptic splice mutations are functional.
Large-scale sequencing studies of patients with autism spectrum disorders and severe intellectual disability have demonstrated the central role of de novo protein-coding mutations (missense, nonsense, frameshift, and essential splice dinucleotide) that disrupt genes in neurodevelopmental pathways. To assess the clinical impact of noncoding mutations that act through altered splicing, we applied the neural network to predict the effects of de novo mutations in 4,293 individuals with intellectual disability from the Deciphering Developmental Disorders cohort (DDD)8, 3,953 individuals with autism spectrum disorders (ASD) from the Simons Simplex Collection and the Autism Sequencing Consortium9-11, and 2,073 unaffected sibling controls from the Simons Simplex Collection. We show that de novo mutations that are predicted to disrupt splicing are significantly enriched in intellectual disability and autism spectrum disorder cohorts compared to healthy controls (Figure 5A in SpliceAI paper). Based on the excess of de novo mutations in affected versus unaffected individuals, cryptic splice mutations are estimated to comprise about 11% of pathogenic mutations in autism spectrum disorder, and 9% in intellectual disability, after adjusting for the expected fraction of mutations in regions that lacked sequencing coverage or variant ascertainment in each study.
We next experimentally validated the accuracy of our cryptic splice site prediction. For this, we obtained peripheral blood-derived lymphoblastoid cell lines (LCLs) from 36 individuals from the Simons Simplex Collection, which harbored predicted de novo cryptic splice mutations in genes with at least a minimal level of LCL expression; each individual represented the only case of autism within their immediate family. As is the case for most rare genetic diseases, the tissue and cell type of relevance (presumably developing brain) was not accessible. Hence, we performed high-depth mRNA sequencing to compensate for the weak expression of many of these transcripts in LCLs. After excluding 8 individuals who had insufficient RNA-seq coverage at the gene of interest, we identified unique, aberrant splicing events associated with the predicted de novo cryptic splice mutation in 21 out of 28 patients. These aberrant splicing events were absent from the other 35 individuals for whom deep LCL RNA-seq was obtained, as well as the 149 individuals from the GTEx cohort. Seven cases did not show aberrant splicing in LCLs, despite adequate expression of the transcript. Although a subset of these may represent false positive predictions, some cryptic splice mutations may result in tissue-specific alternative splicing that is not observable in LCLs under these experimental conditions.
Deep learning is a relatively new technique in biology, and is not without potential trade-offs. Deep learning models can utilize sequence determinants not well-described by human experts, but there is also the risk that the model may incorporate features that do not reflect the true behavior of the spliceosome. These confounding features could increase the apparent accuracy of predicting annotated exon-intron boundaries, but would reduce the accuracy of predicting the splice-altering effects of arbitrary sequence changes induced by genetic variation. Because accurate prediction of variants provides the strongest evidence that the model can generalize to true biology, we provide corroborating evidence of predicted splice-altering variants using three fully orthogonal methods: RNA-seq, natural selection in human populations, and enrichment of de novo variants in case vs control cohorts. While this does not fully preclude the incorporation of irrelevant features into the model, the resulting model appears faithful enough to the true biology of splicing to be of significant value for practical applications such as identifying cryptic splice mutations in patients with genetic diseases.
Our understanding of how mutations in the noncoding genome lead to human disease remains far from complete. The discovery of likely penetrant de novo cryptic splice mutations in childhood neurodevelopmental disorders demonstrates that whole genome sequencing coupled with improved interpretation of the noncoding genome can benefit patients with severe genetic disorders. Cryptic splice mutations have also been shown to play major roles in cancer, and recurrent somatic mutations in splice factors have been shown to produce widespread alterations in splicing specificity12. Much work remains to be done to understand regulation of splicing in different tissues and cellular contexts, particularly in the event of mutations that directly impact proteins in the spliceosome. In light of recent advances in oligonucleotide therapy that could potentially target splicing defects in a sequence-specific manner13, greater understanding of the regulatory mechanisms that govern this remarkable process could pave the way for novel candidates for therapeutic intervention.
Acknowledgements
We would like to acknowledge J. K. Pritchard for insightful discussions and support, the Genome Aggregation Database (gnomAD), and the groups that provided exome and genome variant data to this resource. Stephan J. Sanders was supported by a grant from the Simons Foundation (SFARI #402281 and #574598).
References
- Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880-1887.
- Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209.
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2-3):377-394.
- Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806.
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015;348:648–660.
- Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443.
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433-438.
- De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209-215.
- Sanders SJ, He X, Willsey AJ, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215-1233.
- Turner TN, Hormozdiari F, Duyzend MH, et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet. 2016;98(1):58-74.
- Jung H, Lee D, Lee J, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet. 2015;47(11):1242-1248.
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723-1732.