Next-generation Sequencing Glossary
NGS glossary
Guide to NGS terminology
Use our next-generation sequencing glossary to view definitions of key terms and clarify important concepts as you plan your sequencing project.
The critical difference between Sanger sequencing and NGS is sequencing volume. While the Sanger method only sequences a single DNA fragment at a time, NGS is massively parallel, that is, sequencing millions of fragments simultaneously per run. This process translates into sequencing hundreds to thousands of genes at one time. NGS also offers greater discovery power to detect novel or rare variants with deep sequencing.
Glossary of common NGS terms
Short sequence-specific oligos ligated to the 5ʹ and 3′ends of each DNA fragment in a sequencing library as part of NGS library preparation. The adapters are complementary to the short sequences present on the surface of Illumina flow cells.
The process of matching sequencing reads to a reference genome.
An amplification reaction that occurs on the surface of an Illumina flow cell. During flow cell manufacturing, the surface is coated with a lawn of two distinct oligonucleotides often referred to as “P5” and “P7.” In the first step of bridge amplification, a single-stranded sequencing library (with complementary adapter ends) is loaded into the flow cell. Individual molecules in the library bind to complementary oligos as they “flow” across the oligo lawn. Priming occurs as the opposite end of a ligated fragment bends over and “bridges” to another complementary oligo on the surface. Repeated denaturation and extension cycles (similar to PCR) result in localized amplification of single molecules into millions of unique, clonal clusters across the flow cell. This process, also known as “clustering,” occurs in an onboard cluster module within an NGS instrument.
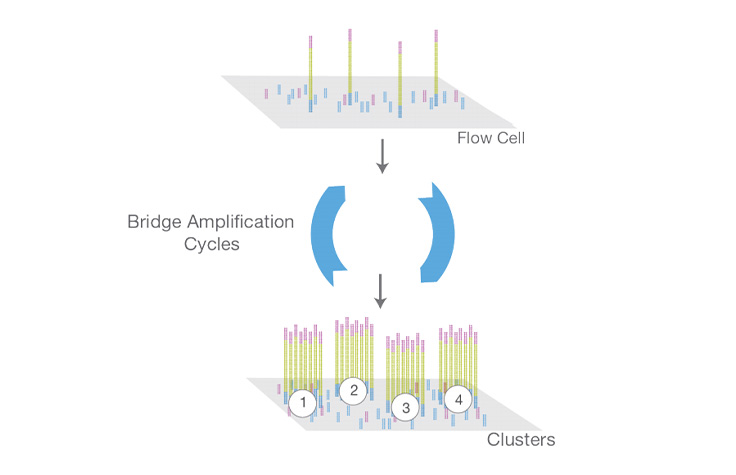
Cluster Amplification
Library is loaded into a flow cell and the fragments are hybridized to the flow cell surface. Each bound fragment is amplified into a clonal cluster through bridge amplification.
A clonal grouping of template DNA bound to the surface of a flow cell. Each cluster is seeded by a single template DNA strand and is clonally amplified through bridge amplification until the cluster has ~1000 copies. Each cluster on the flow cell produces a single sequencing read. For example, 10,000 clusters on the flow cell would produce 10,000 single reads and 20,000 paired-end reads.
Clusters
Pairs of indexes such that every i5 index would be paired with each i7 index in a matrix to create unique index pairs, but not unique single-sided indexes.
A stretch of continuous sequence, in silico, generated by aligning overlapping sequencing reads.
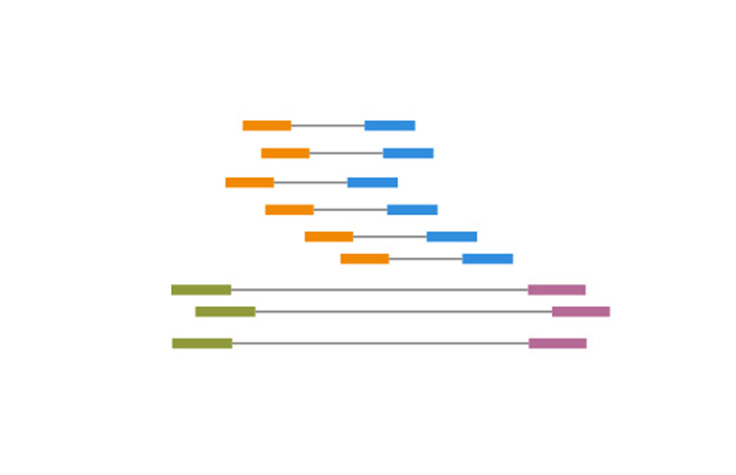
Contigs
The average number of sequenced bases that align to, or “cover,” known reference bases. For example, a whole genome sequenced at 30× coverage means that, on average, each base in the genome was sequenced 30 times. At higher levels of coverage, base calls can be made with a higher degree of confidence.
Learn MoreA metric describing the percentage of bases sequenced across the genome or target region at a given depth (eg, 95% of bases covered with a minimum 10× coverage). Average or mean sequencing depth by itself (eg, 30× mean coverage) does not take into account the percentage of bases sequenced below acceptable threshold limits or bases that were not sequenced at all. For example, when a data set has a reported "coverage distribution of 95% with a minimum 10× coverage," this also indicates that 5% of bases were covered below the 10× threshold or not covered at all. For this reason, coverage distribution is commonly used along with mean coverage to describe sequencing results.
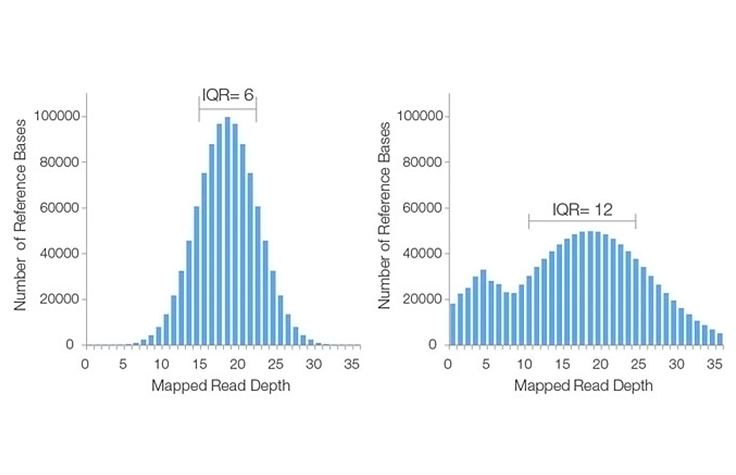
Coverage Distribution
A glass slide or other solid surface with one, two, or eight physically separated lanes used as a consumable on NGS instruments. Sequencing templates are immobilized on the flow cell surface, which is designed to present the DNA in a manner that facilitates access to enzymes while ensuring high stability of surface-bound template and low nonspecific binding of fluorescently labeled nucleotides. Solid-phase amplification (cluster generation) creates up to 1000 identical copies of each single template molecule in close proximity. Densities on the order of ten million clusters per square centimeter are achieved.

A unique short DNA sequence added to each DNA fragment during library preparation. The unique sequences allow many libraries to be pooled together and sequenced simultaneously. Sequencing reads from pooled libraries are identified and sorted computationally, based on their barcodes, before final data analysis. Library multiplexing is a useful technique when working with small genomes or targeting genomic regions of interest. Multiplexing with barcodes can exponentially increase the number of samples analyzed in a single run, without drastically increasing run cost or run time.
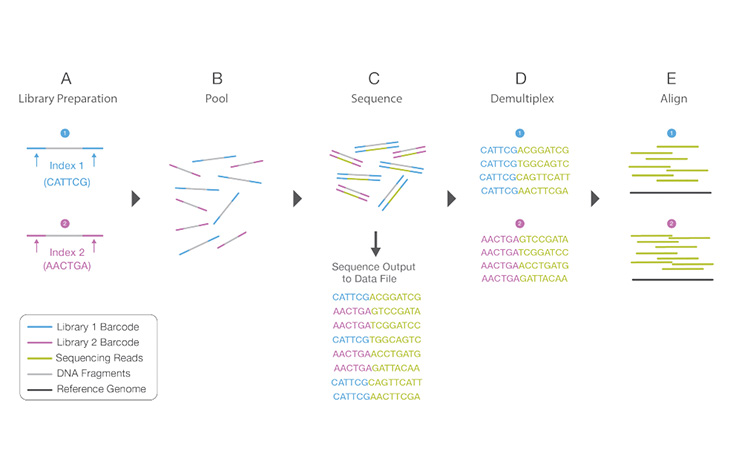
Library Multiplexing Overview
(A) Unique index sequences are added to two different libraries during library preparation. (B) Libraries are pooled together and loaded into the same flow cell lane. (C) Libraries are sequenced together during a single instrument run. All sequences are exported to a single output file. (D) A demultiplexing algorithm sorts the reads into different files according to their indexes. (E) Each set of reads is aligned to the appropriate reference sequence.
i5 and i7 sequencing are the index reads of an Illumina sequencing run, found upstream (i5) and downstream (i7) from the R1 and R2 reads that contain the sequence of interest. Short adapters are added to nucleotide fragments to allow for compatibility with an Illumina sequencer. These sequences bind complementary sequences on a sequencer's flow cell.
During library preparation, sample DNA is fragmented, and the fragments of a specific size (typically 200–500 bp, but can be larger) are ligated or “inserted” in between two oligo adapters. The original sample DNA fragments are also referred to as “inserts.”
A molecular biology protocol that converts a genomic DNA sample (or cDNA sample) into a sequencing library, which can then be sequenced on an NGS instrument. The first step in library preparation is random fragmentation of the DNA sample, followed by ligation of 5ʹ and 3′ adapters to each DNA fragment. Alternatively, “tagmentation” combines the fragmentation and ligation reactions into a single step and greatly increases the efficiency of the library preparation process.
Learn MoreAnother name for next-generation sequencing.
A process by which unique short DNA sequences, or “indexes,” are added to each DNA fragment during library preparation. The unique sequences allow many libraries to be pooled together and sequenced simultaneously. Sequencing reads from pooled libraries are identified and sorted computationally before final data analysis. Library multiplexing is a useful technique when working with small genomes or targeting genomic regions of interest. Multiplexing can exponentially increase the number of samples analyzed in a single run, without drastically increasing run cost or run time.
Learn More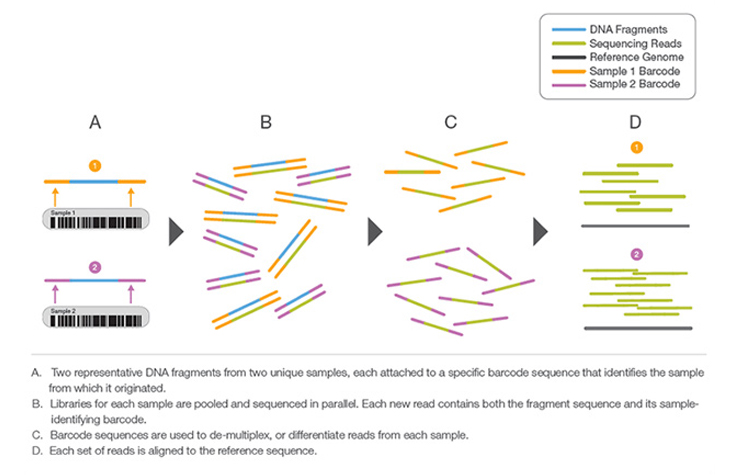
Multiplexing
Having equal proportions of A, C, G, and T nucleotides at each base position across all the DNA fragments in a sequencing library. Color balance is required for effective image analysis on Illumina sequencing systems. Therefore, most Illumina library preparation workflows include a random fragmentation step, which generates the necessary sequence diversity at each base position in the library.
Illumina Knowledge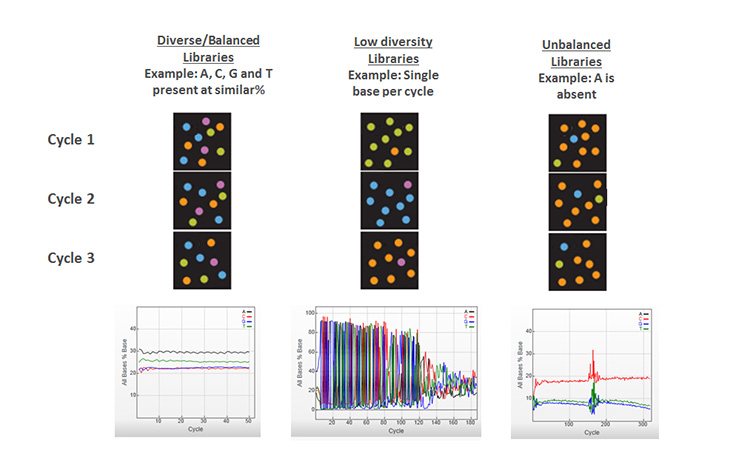
Nucleotide Diversity
A short DNA or RNA sequence.
A process of sequencing from both ends of a DNA fragment in the same run.
Learn More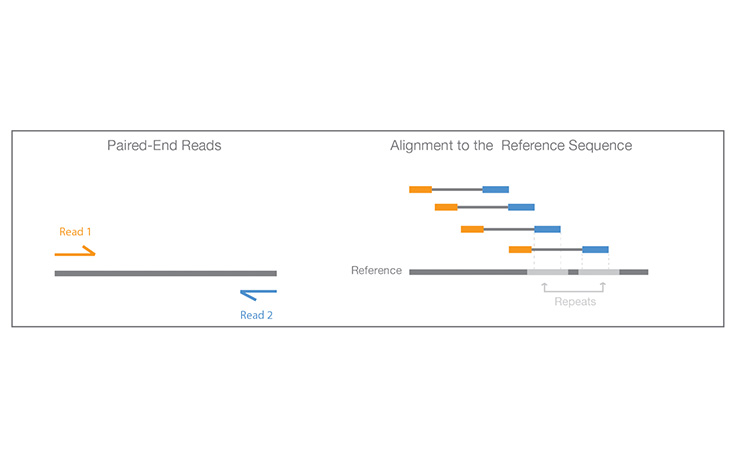
Paired-End Sequencing and Alignment
Paired-end sequencing enables both ends of the DNA fragment to be sequenced. Because the distance between each paired read is known, alignment algorithms can use this information to map the reads over repetitive regions more precisely. This results in better alignment of reads, especially across difficult-to-sequence, repetitive regions of the genome.
A metric in NGS that predicts or estimates the probability of an error in base calling. A quality score (Q-score) serves as a compact way to communicate very small error probabilities. A high Q-score implies that a base call is more reliable and less likely to be incorrect.
Learn MoreOne "read" refers to the forward and reverse sequence of a nucleotide fragment obtained via the bridge amplification process on Illumina flow cells.
A reference genome is a fully sequenced and assembled genome that acts as a scaffold against which new sequence reads are aligned and compared. Typically, reads generated from a sequencing run are aligned to a reference genome as a first step in data analysis. Examples of reference genomes include hg19 and hg38.
SBS technology uses fluorescently labeled nucleotides to sequence the tens of millions of clusters on the flow cell surface in parallel. During each sequencing cycle, a single labeled deoxynucleotide triphosphate (dNTP) is added to the nucleic acid chain. The nucleotide label serves as a “reversible terminator” for polymerization: after dNTP incorporation, the fluorescent dye is identified through laser excitation and imaging, then enzymatically cleaved to allow the next round of incorporation. Base calls are made directly from signal intensity measurements during each cycle. Because all four reversible terminator-bound dNTPs (A, C, T, G) are present as single, separate molecules, natural competition minimizes incorporation bias.

Overview of Illumina Sequencing by Synthesis Workflow | XLEAP SBS chemistry
Refers to the ability to detect a particular variant in a given sample. The lower the allele frequency, the higher the sensitivity needed to detect it. Next-generation sequencing provides higher sensitivity than capillary electrophoresis, offering the ability to detect rare mutations.
A PCR primer adjacent to an adapter sequence that indicates the starting point of the sequencing read. During the sequencing process, the primer anneals to a portion of the sequencing adapter on the template strand. The DNA polymerase enzyme binds to this site and incorporates complementary nucleotides base by base into the growing opposite strand.
The data strings of A,T, C, and G bases corresponding to each DNA fragment in a sequencing library. In Illumina technology, when a library is sequenced, each DNA fragment produces a cluster on the surface of a flow cell and each cluster generates a single sequencing read. (For example, 1 million clusters on a flow cell would produce 1 million single reads and 2 million paired-end reads.) Read lengths can range from 25 bp to 300 bp or higher depending on application needs.
The brightness of the fluorophores that are attached to a substrate. In sequencing by synthesis, base calls are made directly from signal intensity measurements during each cycle.
Illumina supports several indexing methods, including single and dual indexing. With single indexing, up to 48 unique 6-base indexes can be used to generate up to 48 uniquely tagged libraries. With dual indexing, up to 24 unique 8-base Index 1 sequences and up to 16 unique 8-base Index 2 sequences can be used in combination to generate up to 384 uniquely tagged libraries.
A rapid enzymatic reaction where double-stranded DNA is simultaneously fragmented and tagged with Illumina adapter sequences and PCR primer binding sites. The combined reaction eliminates the need for a separate mechanical shearing step during library preparation.
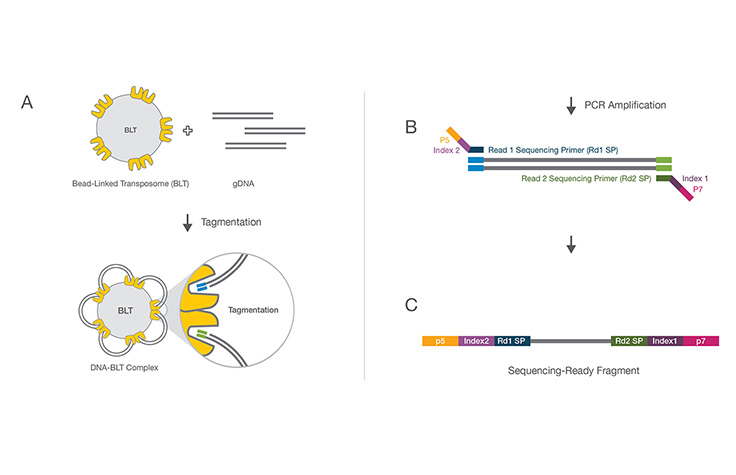
Tagmentation
The total size of all the target regions in a targeted resequencing experiment. Target sizes will vary among different predesigned panels or custom panels. (For example, a panel of 22 target regions, where each target region is 100 kb, will have a total target size of 2200 kb.)
The amount of data produced by a next-generation sequencing instrument. Usually defined in terms of megabases (Mb) or gigabases (Gb).
1 megabase = 1,000,000 bases.
1 gigabase = 1,000,000,000 bases.
1 megabase = 1,000,000 bases.
1 gigabase = 1,000,000,000 bases.
Pairs of indexes such that every i5 index and every i7 index are used only one time. With unique dual indexes, you can identify and filter indexed hopped reads, which provides higher confidence in multiplexed samples.
Read BulletinA widely used sequencing method that targets only the protein-coding region of the genome (the exome).
Learn MoreA comprehensive method that provides a base-by-base view of the entire genome.
Learn More
Getting Started with NGS eBook
Curious about using NGS for your research? While adopting a new technology may seem intimidating, we’ve created a comprehensive, yet easy-to-follow guide for bringing NGS into your lab. You’ll learn about NGS methods, workflows, data analysis solutions, additional glossary terms, and find a step-by-step guide to getting started with NGS.
Download eBookDownload the New to NGS eBook
Additional resources
NGS for beginners
Want to learn more about NGS? This page offers simple, clear explanations of next-generation sequencing and its numerous benefits over conventional methods.
NGS tutorials
Browse our comprehensive list of tutorials designed to help you understand key concepts in NGS. We’ll guide you through best practices for library prep, sequencing, and data analysis.
Online training
Access on-demand training for instruments, library prep kits, software, and more.