
Cancer Research Methods Guide
In this methods guide, get an overview of Illumina sequencing followed by workflow suggestions for popular cancer research methods.
Cancer is a disease of the genome, but genetic mutations are only one factor. Nongenetic changes also affect phenotypes. Understanding the epigenetic landscape of cancer is increasingly crucial for modeling cancer initiation, progression, and therapeutic responses. Cancer epigenetics studies can uncover how cells control gene activity through processes such as DNA methylation.
Epigenomic technologies can identify cellular biomarkers associated with regulation of cancer genes or drug resistance:
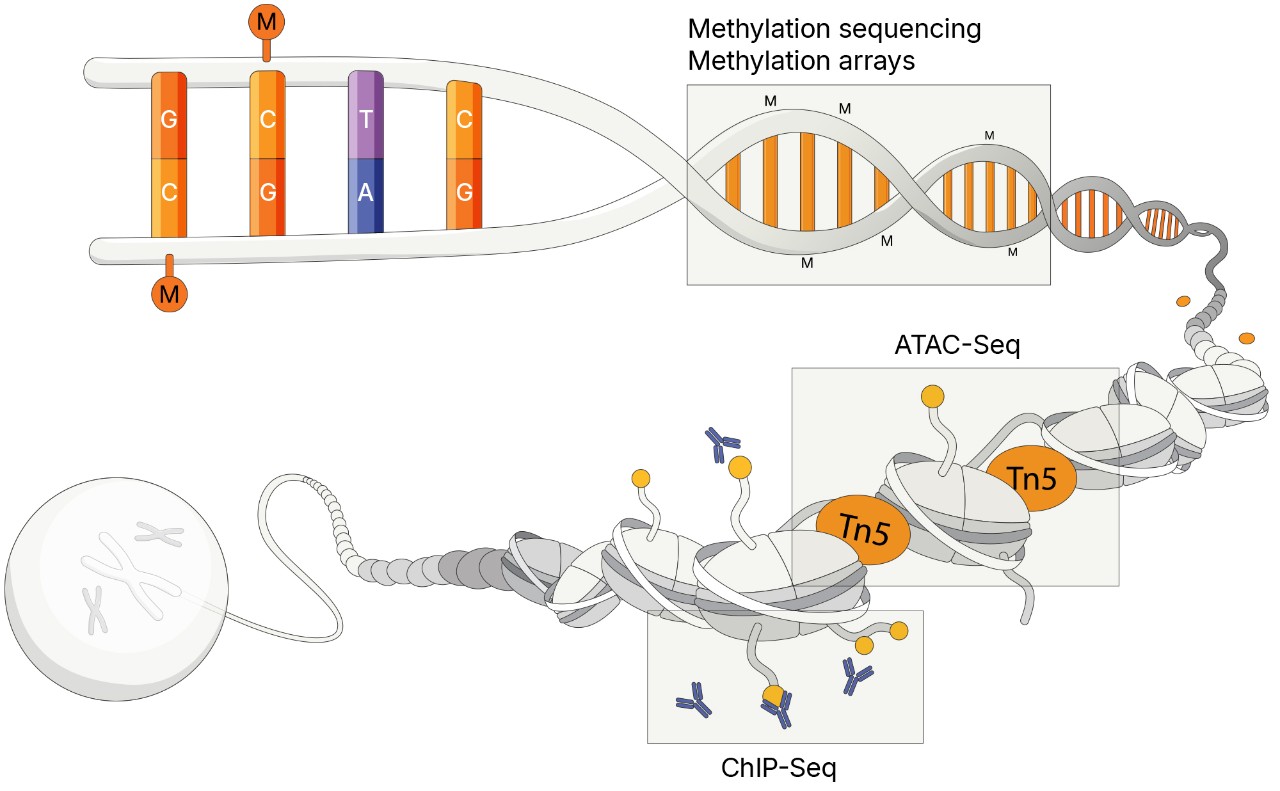
Methylation sequencing provides a powerful approach for studying epigenetic changes in cancer, offering single-base resolution of DNA methylation patterns that influence gene regulation and tumor progression. Using next-generation sequencing (NGS), researchers can profile methylation events associated with oncogenesis. Certain methods can detect both genetic variants and methylation patterns in a single workflow, enabling simultaneous genomic and epigenomic insights critical for identifying somatic mutations and aberrant methylation signatures.
Learn more about:
Detects methylation at cytosines across the genome with whole-genome methylation sequencing
Captures emerging regions of interest, including enhancers and transcription factor binding sites
Requires only minimal DNA input, making it suitable for cancer samples, such as liquid biopsies
Enables analysis of cell-free DNA (cfDNA) samples for noninvasive cancer detection and monitoring studies
Methylation arrays allow researchers to quantitatively interrogate methylation across the epigenome of cancer cells at single-site resolution. Array-based methylation profiling delivers highly accurate, targeted interrogation of CpG islands, selected CHH sites, enhancers, open chromatin regions, and transcription factor binding sites.* As a high-throughput research method, arrays have a lower cost per sample compared with alternatives.
Methylation array protocols have a user-friendly, streamlined workflow with > 98% assay reproducibility and support for formalin-fixed, paraffin-embedded (FFPE) samples, increasing the applicability of methylation arrays to biobanked tissues.
*CpG: Genomic region where cytosine is directly followed by guanine; CHH: Other genomic regions where methylation can occur, defined by a cytosine that is immediately followed by any nucleotide other than guanine (H = A, T, or C)
Offers cost-effective methylation profiling
Delivers highly precise methylation measurements at targeted sites
Enables highly scalable sample processing
Provides simple data analysis with low computational burden
Cancer researchers can use the assay for transposase-accessible chromatin with sequencing (ATAC-Seq) to study epigenetic features across the genome without prior knowledge of regulatory elements. ATAC-Seq exposes genomic DNA to Tn5, a highly active transposase that preferentially inserts into open chromatin sites and adds sequencing primers. Subsequent NGS analysis, which can include genomic or transcriptomic profiling, provides insights into chromatin accessibility across the genome.
Uncovers open chromatin across the genome without predefined targets for unbiased discovery
Interrogates precious cancer samples with lower sample input than FAIRE-Seq and DNase-Seq
Produces rapid results with a streamlined workflow
Complements RNA-Seq studies that can directly determine if regions of open chromatin are being expressed
Chromatin immunoprecipitation sequencing (ChIP-Seq) is a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. This method can reveal insights into gene regulatory events and biological pathways that play important roles in the development and progression of some cancers. Illumina offers efficient workflow solutions to enable genome-wide surveys of gene regulation using ChIP-Seq.
Captures DNA targets for transcription factors or histone modifications across the genome
Defines transcription factor binding sites
Reveals gene regulatory networks when combined with RNA-Seq and methylation analysis
Offers compatibility with various input DNA samples
Cancer Research Methods Guide
In this methods guide, get an overview of Illumina sequencing followed by workflow suggestions for popular cancer research methods.

Decoding complex pathways with 5-base analysis for multiomic insights
In this eBook, discover the value of studying the methylome and genome, together, to unravel the mechanisms of genetic disease, cancer, and other complex diseases.

Liquid biopsy eBook
This 20+ page eBook provides published, comprehensive workflows for thorough characterization of liquid biopsy samples using NGS and microarrays.

Robust methylation profiling microarray providing extensive coverage of CpG islands, genes, and enhancers. Ideal for genetic and rare disease research, cancer research, and classification.

A single assay for comprehensive discovery of the 5-base genome (A, T, G, C, and 5mC), providing dual insights into the whole genome and methylome.

A single assay for targeted detection of five DNA bases (A, T, G, C, and 5mC), providing dual insights into genomic variants and methylation events.

Importance of epigenetics in cancer research
This infographic about epigenetics serves as a visual guide to see why nongenetic changes greatly impacts the understanding of cancer.

Understanding cancer biology with methylation microarrays
In this video, cancer biology experts discuss the many advantages of leveraging methylation microarrays for biomedical research.

Advancing cancer research with multiomics
In this video, learn how to link the causes and consequences of complex phenotypes through multiomics to enable discoveries that weren’t possible before.
Interested in learning more about methods to study cancer epigenetics?